论文解读<<基于分布式特征表示的深度学习模型识别蔷薇科基因组中的DNA n4 -甲基胞嘧啶位点>>
论文解读<<基于分布式特征表示的深度学习模型识别蔷薇科基因组中的DNA n4 -甲基胞嘧啶位点>>
摘要
DNA n4 -甲基胞嘧啶(4mC)是在原核生物和真核生物中发现的一种表观遗传修饰,涉及许多生物学功能,包括宿主防御、转录调节、基因表达和DNA复制。为了识别4mC站点,以前的计算研究主要集中在寻找手工制作的特征。因此,这一研究领域将受益于计算机方法的发展,该方法依赖自动特征选择来识别相关的地点。本文报道了4mC-w2vec,一种基于分布式特征表示,通过单词嵌入技术“word2vec”学习自动识别蔷薇基因组,特别是蔷薇(rosa chinensis, R. chinensis)和fragaria vesca (F. vesca)特征的计算方法。虽然目前使用一些生物信息学工具来识别这些基因组中的4mC位点,但它们的预测性能不充分。我们的系统通过单词嵌入过程处理4mC和非4mC站点,通过k-mer处理其生物词的子词信息,将子词信息作为特征输入到双层卷积神经网络(CNN)中,以分类样本序列是否包含4mC站点或非4mC站点。
1、介绍
表观遗传学是指与DNA序列本身[1]的修饰无关的基因功能的可遗传变化。DNA甲基化是最广为人知的表观遗传标记之一,因为它起着至关重要的作用在各种重要的生物学过程,包括改变染色质结构,确保稳定的DNA,基因表达控制DNA构象,x染色体失活、基因调控、细胞分化、和癌症进展[2 - 5]。最广泛的DNA甲基化修饰之一是n4 -甲基胞嘧啶(4mC),它是主要在1983年描述的[6],它在真核生物和原核生物的胞嘧啶环的第4位被甲基化(尽管4mC在后者中更常见和研究)。在原核生物中,4mC是一个限制修饰(R-M)系统的一部分,该系统可以防御外来DNA的活动,包括其修复、表达和复制[7-11]。4mC在基因组稳定、重组和进化等方面也起着辅助作用[12-14]。4mC在真核生物中的生物学作用尚不清楚,部分原因是真核生物基因组中4mC的体积很小,因此无法通过高灵敏度技术以外的任何方法检测到它。为了实验识别4mC位点,单分子实时荧光定量PCR (Single Molecule of RealTime, SMRT)、质谱和甲基化精确PCR都被使用了[15-18]。但是,****这些方法都是费时费力的。利用适当的计算工具分析与山茱草基因组相关的“大数据”,可能是准确识别4mC位点的更有效手段目前只有两种计算方法可以识别蔷薇科基因组中的4mC位点:i4mC-Rose[33]和DNC4mC-Deep[34]。i4mC-Rose工具是具有多种编码方案的随机森林分类器的结果,而DNC4mC-Deep是具有六种编码技术的深度学习方法的结果。尽管这些方法产生了可接受的结果,但仍然有很大的改进空间,特别是考虑到采用的数据集可能没有足够的质量来捕获4mC基模,或者所采用的特征选择方法可能不适合区分正类和负类的序列信息。此外,以往的方法依赖领域知识对输入特征进行手工设计。相比之下,我们的方法通过单词嵌入自动捕获高水平的输入特征,允许一个新颖和高度精确的计算工具。本文开发了基于序列的DNA 4mC位点预测器。我们的核心思想是通过单词嵌入将DNA序列转化为向量,然后用双层一维CNN对其进行处理,最终进行分类。字嵌入谷歌于2013年诞生在应用[35]协助自然语言处理(NLP),但后来发现成功的生物应用程序(第36 -),深度学习的使用在我们的第二步在许多方面取得了显著的结果,包括语音识别[44],图像识别(45、46),NLP[47]和全基因组预测[48-53]。在我们的研究中,结合词嵌入技术和深度学习技术,在平衡和不平衡类数据集上都取得了出色的结果,我们认为所提出的方法在全基因组预测中是有前景的。
2、材料和方法
2.1、数据集组成
为了开发基于序列的标识符,有必要构建可靠的数据集。我们独立地构造了一个完整的训练集和独立的数据集。从MDR数据库[54],http://mdr.xieslab.org/中获得4mC序列(阳性序列)。根据以往的研究,长度为41 nt的预测效果最好[22,29]。因此,DNA序列的长度被设置为41 nt,在中心包含‘C’。以往的研究者[33,34]使用P20的修正QV (modQV)评分来生成正数据集,但w.c chen等人指出,modQV评分为30或30以上是将胞嘧啶位置标记为modified[21]的默认或最佳阈值。为了开发更可靠的模型,我们应用了p30的QV来构建我们的正数据集,并排除了QV值<30的序列。为了去除序列相似性,使用CD-HIT[55]软件,截断阈值为65.00%。结果,我们获得了4321个vesca基因组,2421个inR阳性序列。chinensisgenome。从这些数据集中,选择约80%的序列(3457 (F. vesca)和1938 (R. chinensis))作为训练集,剩余序列(864 (F. vesca)和483 (R. chinensis))作为独立数据集。
阴性序列(非4mC位点序列)来自同一基因组文件,其中4mC位点(位于中心的“C”)未被SMRT测序技术检测到。这样,在每个物种中都形成了以“C”为中心的大量负序列。在模型训练中,正序列和负序列被平衡。为了测试效果基于该模型,我们构建了具有不同正负样本比率的独立数据集。ForF。Vescathese分别为:1:1[864阳性864阴性],1:5[864阳性4320阴性],1:15[864阳性12960阴性]。中华绒螯蟹:1:1[阳性483条,阴性483条],1:5[阳性483条,阴性2415条],1:15[阳性483条,阴性7245条]。由于独立阳性序列的数量有限,所有比值组(即864 forF)均接受相同的阳性序列。要使vescaand 483。对)。阴性序列在比值组之间没有重叠。表1总结了这两个物种的训练和独立数据集。

2.2、方法
我们提出了一种新的方法(4mC-w2vec)来鉴定蔷薇科基因组中的4mC位点。我们的计划包括两个主要步骤。第一步是鉴别特征生成或表示阶段,该阶段将每个DNA序列用3-mer描述成单词,然后使用单词嵌入方法将每个单词映射到对应的特征表示。第二步,利用深度学习模型,根据第一阶段生成的特征对4mc和非4mc进行分类。下面几节将给出详细的解释,并在fig中给出通用的体系结构。1.

2.2.1。分布式特性表征
我们决定应用一种通常被称为“word2vec”[35]的单词嵌入技术。该技术基于分布假设[56]生成最优特征向量集。Word2vec是一个两层神经网络,它通过向图中所示的单词向量化来处理文本。1(一)。它以文本语料库的形式接收输入,输出是表示语料库中单词的特征向量。这种技术降低了计算复杂度,降低了噪声,最终提高了计算模型的性能。此外,许多生物密码(如遗传密码)可以表示为一种语言[57-59],由此产生的见解可以用于解决各种生物学问题[58,60,61]。因此,我们采用word2vec方法来寻找每个4mC位点的可解释表示。
语料库的构建发现了词语大文件之间的语义关系。在我们的研究中,我们通过生成了语料库。F. vescaand和R. chinensis利用NCBI基因组数据生成了语料库,可通过网址://www.ncbi.nlm.nih.gov。训练word2vec的第一步是建立语料库词汇。word2vec模型可以基于连续词袋法(CBOW)或跳跃图法应用。在跳跃图模型中,使用当前单词(w(t))或输入来预测上下文单词的周围窗口。相比之下,CBOW方法尝试根据邻近的单词(上下文)猜测目标单词。作为CBOW模型的输入,窗口大小为5的公式如下seqn 1:
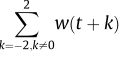
CBOW和Skip-gram的表现类似,尽管Skipgram更有用,对不常见的单词给出更好的结果[62]。在我们的研究中,我们关注的是频繁的单词,因此采用CBOW进行word2vec训练。为了处理CBOW,基因组组装被分成长度为200 nt的句子。接下来,每个句子被分成重叠的3-mer组成单词(如AAT, CCT, GCN, CCC)。在这一点上,每个4mC包含一条连续的核苷酸链。这些单词被输入到一个两层的word2vec模型中,如图1(a)所示。因此,每个单词都有自己的100维(D)向量表示,每个lengthl序列由一个shapeðL?2Þ? 100。例如,单词“AAT”被表示为100-(D)向量的½0:111;0:222;0:333;…::;0:12100?和’ CCT '表示为100-D向量的½0:221;0:112;0:313;…::;0:23100?表2列出了用于训练word2vec的参数。大多数参数保持默认值。根据前人的研究,通过创建的方法获得了最佳的性能****100 - d(36, 43,50)。因此,将单词向量的维数参数设为100-D。我们包括所有频率大于1的单词。对于上下文词,我们测试了不同的重叠k-mers,如k = 1 (A), k = 2 (AT), k = 4 (ATCG), k = 5 (ATCGA),和k = 6 (ATCGAT)。负采样设置为5,以绘制“噪声词”。窗口大小设置为5,表示句子中当前单词和预测单词之间的最大距离。语料库上的纪元(迭代)数设置为20。word2vec是使用python库genism对两个物种独立训练的[63]。

2.2.2。CNN模型
我们使用CNN模型(深度前馈神经网络)学习word2vec生成的特征。在CNN中,超参数决定了训练步骤中的层结构,这影响了模型的准确性和学习时间。因此,采用网格搜索策略进行超参数优化,包括滤波器个数、核大小(滤波器大小)、退出率、卷积层数和激活函数。在应用网格搜索技术后,所提出的CNN模型生成了两个一维卷积层,包含64个9单元的滤波器和一个跨单元。在每个卷积层中,使用一个整流线性激活单元(ReLU)作为激活函数。为了解决过拟合问题,第一层卷积之后是一个辍学率为0.7的辍学率层。对于最终的分类,使用了一个全连接层,其中一个节点,后面是s形函数。CNN模型的配置如表3所示。为了在训练数据集中训练CNN模型,设置学习率为0.0007,批次大小设置为128,并基于验证损失采取早期停止策略。采用RMSprop作为优化器[64],采用二元交叉熵作为损失函数[65]。Keras框架(一个python开源库,https://keras.io/)被用于构建4mC-w2vec。经过训练的模型将能够通过在CNN训练阶段设置“类权重”来学习不平衡的类数据集。

2.3. 评价参数
使用各种统计指标,包括敏感性(Sn)、特异性(Sp)、准确性(ACC)和马修相关系数(MCC)来评估模型的性能[67-69]。算式中TP、FP、TN、FN为真阳性、假阳性、真阴性、假阴性值。我们还纳入了受试者工作特征(ROC)曲线来评价所提出的方法。

3.结果和讨论
3.2. 使用不同编码方法的效果
基于k-mer值(如1-mer、2-mer、3 -mer、4-mer、5-mer和6-mer)的重叠,通过词嵌入得到6个特征向量模型。将这些向量表示模型输入CNN进行4mC站点的独立识别。我们观察到,3-mer在预测两种物种的4mC位点方面提供了更多的信息。在这项研究中,基于3-mer并被CNN分类的word2vec表示被认为是最终模型,或“i4mC-w2vec”。在交叉验证检验中,F. vesca预测因子的MCC为0.7407,精度为0.8697,AUC为0.9400。。R. chinensis.的MCC为0.7093,精度为0.8541,AUC为0.9370根据以往的研究,在4mC预测任务[20]中,采用one-hot方法结合深度学习模型编码的生物序列表现良好。我们采用一次热编码方案对DNA序列进行编码,其中A、C、G、T核苷酸分别编码为(1 0 0 0)、(0 1 0 0)、(0 0 1 0)和(0 0 0 1)。为了确定单热编码CNN的最佳参数,采用了网格搜索算法。结果表明,基于3-mer和4 -mer的word2vec方法优于一次性热法。表4给出了CNN分类时基于不同k-mers和一次热编码的六词嵌入模型的性能。更普遍地说,f。采用单热编码的vescais为0.8920,采用word2vec (3-mer)编码的vescais为0.9400(图3)(a)。chinensis在一次热编码时为0.9110,在word2vec (3-mer)时为0.9370(图3)(b)。
3.3. 与已有方法在独立测试数据集上的性能比较


