利用三级结构进行蛋白质嵌入的自我监督预训练
文章目录
- 一、摘 要
- 二、Introduction
- 三、 Related Work
-
- 3.1 蛋白质三维结构依赖的任务
- 3.2 自我监督学习
- 四、 Methods
-
- 4.1 蛋白质结构的SE(3)-不变表示
- 4.2 自我监督预训练
- 4.3 下游任务的预训练模型
- 五、总结
- 六、Experiments
-
- 6.1 下游任务的预训练模型
- 七、Results
- 八、Conclusion
一、摘 要
蛋白质的三级结构在很大程度上决定了它与其他分子的相互作用。尽管它在各种与结构相关的任务中很重要,但完全监督的数据往往耗时且获取成本高。现有的训练前模型主要关注氨基酸序列或多序列比对,而结构信息尚未得到利用。在本文中,我们提出了一个自监督的预训练模型来学习结构嵌入从蛋白质三级结构。原生蛋白质结构受到随机噪声的扰动,而训练前模型的目的是估计被扰动的三维结构上的梯度。具体地说,我们采用SE(3)-不变特征作为模型输入,并以保留SE(3)-等方差的方式重建三维坐标上的梯度。这种范式避免了复杂的SE(3)-等变模型的使用,并显著提高了训练前模型的计算效率。我们证明了我们的预训练模型在两个下游任务上的有效性,蛋白质结构质量评估(QA)和蛋白质-蛋白质相互作用(PPI)位点预测。通过提取层次结构嵌入来增强相应的预测模型。大量的实验表明,这种结构嵌入始终提高了两个下游任务的预测精度。
二、Introduction
蛋白质的生物学功能以及可能与其他分子的相互作用,很大程度上取决于其三维结构(Bergetal.2002)。对于各种蛋白质相关的应用,例如基于结构的药物设计(SBDD)(“雪橇”z和咖啡馆2018;蝙蝠池、Ahmad和Choi2019)和蛋白质相互作用(PPI)预测(Sun等,2017;曾等,2020年),蛋白质三级结构是最关键的特征之一。然而,通过实验结构测定来收集蛋白质-配体复合物和多蛋白质复合物的三维结构是费时费力和昂贵的。因此,SBDD和PPI模型的性能往往受到有限的结构数据的限制。另一方面,蛋白质结构预测的计算方法几十年来已经引起了越来越多的关注。
通过各种预测工具可以生成大量的结构诱饵,这就提出了如何找出最大诱饵的问题准确预测,即蛋白质结构质量评估(QA)(Olechnoviˇc和Venclovas 2017;Baldassarre等人。2021).
由于由不同协议产生的结构诱饵可能具有高度多样化和non-i.i.d.,因此获得蛋白质结构的通用嵌入是至关重要的。总之,蛋白质结构嵌入在许多蛋白质相关应用中至关重要,但由于数据有限和/或数据分布的潜在偏差,获取这些信息并不重要。
自然语言处理(NLP)的最新进展表明,大规模的自我监督预训练模型在各种下游任务中可以非常有效(Vaswani等人,2017年;Devlin等人,2019年)。类似的想法也被用于训练蛋白质的大规模语言模型,包括氨基酸序列或多序列比对(MSAs)。在(Rao等人2019;Rives等人2021)中,训练LSTM和变压器模型预测FASTA序列中随机屏蔽的氨基酸,从而形成蛋白质内残基间的相互作用。Sturmfels等人(Sturmfels等人,2020年)提出预测来自多个序列比对的图谱,而不是随机掩盖的氨基酸。在(Raoetal.2021)中,我们训练变压器模型来预测多个序列比对(而不是FASTA序列)中的掩蔽位置,从而更好地配合msa中嵌入的协同进化信息。所有这些基于序列的预训练模型都已被证明在学习氨基酸类型的有意义的嵌入方面是有效的,并为二级结构和接触预测提供了关键的特征。
然而,这种基于序列的预训练模型并没有利用蛋白质的三级结构,而这可能对上述与结构相关的下游任务至关重要。此外,大规模语言模型的计算复杂度通常非常高,通常需要在高性能GPU集群上训练此类模型还需要数周甚至数月(Raoetal.2021)。
为了解决上述问题,我们提出了一个从蛋白质三级结构中学习结构嵌入的预训练模型。该模型采用自监督损失函数进行优化,该函数仅依赖于蛋白质结构,不需要任何额外的监督。
具体来说,天然蛋白质结构受到高斯噪声的随机干扰,该模型旨在估计受干扰的三维坐标上的对数概率梯度。
由于三维旋转和平移的内在对称性,在梯度估计中必须保持SE(3)-等方差。标准SE(3)-等变模型通常涉及复杂而耗时的球谐计算(Thomas等2018;Fuchs等2020年)或正则表示(哈钦森等2020年)。相比之下,我们构造SE(3)不变特征作为训练前模型的输入,然后在保留SE(3)等方差的情况下重建三维坐标上的梯度。这种工作流程,类似于(Shietal.2021),在不牺牲SE(3)-等方差的情况下,显著提高了计算效率。
我们通过两个下游任务来证明我们的预训练模型的有效性:蛋白质结构质量评估和蛋白质-蛋白质相互作用位点预测。利用预先训练的模型提取层次结构嵌入(全蛋白、残基内和残基间),并将其输入到针对每个下游任务提出的相应模型中作为增强。大量的实验表明,这种结构嵌入可以持续地提高下游任务的预测精度。
本文的总体贡献总结如下:
1、我们提出了第一个蛋白质三级结构的自我监督预训练模型,而现有的模型只利用氨基酸序列或多序列比对。
2、我们的预训练模型的计算效率很高,并且能够在不同的层次上生成信息性的结构嵌入。
3、我们证明了通过我们的训练前模型提供的协同结构嵌入,可以持续提高下游任务的预测精度。
三、 Related Work
3.1 蛋白质三维结构依赖的任务
在本文中,我们采用了两个需要蛋白质三维(3D)结构的下游任务来评估我们的训练前模型:蛋白质模型质量评估(QA)和蛋白质-蛋白质相互作用(PPI)位点预测。
蛋白质结构QA(模型精度的估计)根据计算蛋白质模型与原生结构的差异来估计计算蛋白质模型的质量(Wonetal.2019)。它的目的是1)在蛋白质结构预测模型池中找到最佳模型,2)根据其估计的局部质量细化模型。QA任务利用了两种类型的评估指标:本地分数和全局分数。在残差水平上,局部得分包括局部距离差异检验(LDDT)(Mariani等,2013)和接触面积差异(CAD)(Olechnoviˇc、库伯基特和文克拉瓦斯2013)得分。在蛋白质水平上,全局得分包括全球距离测试总分(GDTTS)(巴尔达萨雷等人,2021年)、全球距离测试高精度(GDTHA)(Zemla,2003年)、tm得分(Zhang和斯科尔尼克,2004年)以及LDDT和CAD的全球版本。
蛋白质-蛋白质相互作用是指两个或多个蛋白质之间的物理接触,这对蛋白质的功能至关重要(DeLasRivas和丰塔尼罗2010a;Zengetal.2020)。PPI位点的鉴定是一种帮助理解蛋白质生物学功能的有效方法(Lietal.2018)。PPI位点预测是一个残留级2状态分类任务。
3.2 自我监督学习
自监督学习方法以其在训练过程中使用大量未标记数据在NLP任务中的良好表现而闻名。它不需要明确的人类指导,也带来了灵活性(Vaswanietal.2017)。一种有效的自监督训练策略是在数据中添加一定的噪声,然后训练网络获得原始数据,这被认为是一个自恢复过程。例如,掩码令牌预测(Devlinetal.2019)用替代令牌替换多个位置的令牌值,并允许网络进行反向预测。最近,一种名为TAPE(Rao的新型蛋白质序列自监督方法atal.2019)使用这种掩蔽标记机制训练预训练模型,并在多个基于序列的预测任务上取得了良好的性能。然而,由于蛋白质三维结构的复杂性,目前还没有基于结构的预训练方法来适应上述三维结构依赖的下游任务。
四、 Methods
在本节中,我们将描述如何用保持SE(3)-不变性来表示蛋白质结构,即对任意的三维旋转和平移具有不变性。之后,我们提出了我们的蛋白质结构的预训练框架,建立在基于能量的模型之上。最后,我们演示了如何将预训练好的模型用于两个下游任务:蛋白质结构质量评估(QA)和蛋白质相互作用(PPI)位点预测。
4.1 蛋白质结构的SE(3)-不变表示
蛋白质的三级结构在很大程度上是由所有氨基酸残基的Cα原子的三维坐标决定的(Gront,Kmiecik,和科林斯基2007;克里沃夫,沙波瓦洛夫,和邓布拉克Jr2009)。因此,用Cα原子的三维坐标来表示蛋白质结构通常就足够了。然而,这种基于坐标的表示取决于蛋白质结构的整体配置(位置和方向)。由于刚体的旋转和平移可以是任意的,并且不会影响蛋白质的结构,因此需要基于协调的模型必须保持SE(3)-等方差,才能在构象空间中捕获这种对称性。在本文中,我们通过引入蛋白质结构的SE(3)-不变表示来规避这种SE(3)-等方差约束。
具体来说,我们计算所有Cα原子对之间的欧氏距离,并用得到的成对距离矩阵表示蛋白质结构。由于相对距离w.r.t.保持不变。任何3D旋转和平移,这种SE(3)不变表示允许对后续模型进行更灵活的选择。

我们的训练前的模型是建立在成对的距离矩阵上的,因此模型本身不需要被约束来保持SE(3)-等方差。然而,值得一提的是,通过链式规则将估计的梯度从成对的距离矩阵传播到三维坐标上是可行的,这对于训练基于能量的模型是至关重要的,我们将在后面进行演示。
4.2 自我监督预训练
为了提取信息性的蛋白质和每个残基嵌入,我们提出了一个预训练模型来近似蛋白质三级结构的数据分布。其内在动机是,如果底层数据分布很好地近似,那么这个训练前模型必须捕获了嵌入在蛋白质结构中的关键信息,这可能对各种下游任务非常有益。
在(Song和Ermon2019)中,Song等人提出通过去噪分数匹配(Vincent2011)来训练一个基于能量的模型用于图像生成。利用不同尺度的高斯噪声对原始图像进行扰动,并训练网络来估计扰动图像上的对数概率梯度。虽然成对距离矩阵作为蛋白质结构的SE(3)不变表示,也可以看作是二维图像,但直接用随机噪声扰动距离矩阵是不合理的。关键的区别在于,对于图像生成任务,每个随机扰动的图像都是有效的,因此扰动的数据分布仍然有很好的定义。然而,并不是所有的L×L重值矩阵都是有效的距离矩阵,即可能不存在满足随机摄动距离矩阵的三维结构。
为了解决这一问题,我们提出了在距离矩阵上的随机扰动,首先在所有Cα原子的三维坐标上添加高斯噪声,并推导出相应的距离矩阵作为摄动输入。然后训练分数网络来估计扰动距离矩阵上的梯度。分数网络的输入和输出对三维旋转和平移都是不变的,因此分数网络可以被任何卷积神经网络实例化。由于随机扰动是在三维坐标上执行的,所以我们在三维坐标上只有封闭形式的地面-真值梯度。因此,我们还需要将估计的梯度从距离矩阵传播到三维坐标,这是通过链规则来实现的。





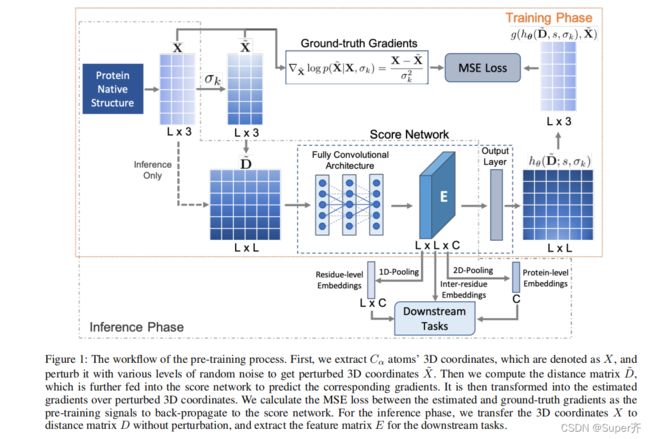
一旦训练前的模型得到了充分的优化,我们就可以利用它来提取新的蛋白质结构的结构嵌入。回想一下,分数网络采用了二维卷积网络作为骨干架构。对于任何特定的蛋白质结构,我们计算所有Cα原子的成对距离矩阵,并将其送入训练前模型。然后提取大小为L×L×C的最终特征图(紧邻估计的梯度),其中C是特征图通道的数量。此外,这些特征图应用一维和二维全局池,通过C维得到了每个残基的结构嵌入和全蛋白质结构嵌入。最后,在推理阶段(如图1所示),我们可以提取全蛋白质、残基内和残基间结构嵌入作为下游任务的额外输入。
4.3 下游任务的预训练模型
在这里,我们以两个下游任务为例,以演示如何利用预训练模型产生的结构嵌入来提高下游任务的预测精度。
蛋白质结构质量评估
由于初始化和优化过程的随机性,大多数蛋白质结构预测方法都生成多个结构诱饵作为相同氨基酸序列的候选对象(Yangetal.2020;Juetal.2021)。蛋白质结构质量评估(QA)旨在在所有候选结构中确定最佳的预测结构,是蛋白质结构预测中不可或缺的模块之一。在(Baldassarreetal.2021)中,作者提出GraphQA将蛋白质结构表示为一个图,其中节点是氨基酸残基,边缘是残基间的相互作用。
为了同时考虑序列结构和几何结构,GraphQA构建了序列相邻和空间相邻残差对的边。该模型由多个消息传递操作组成(Gilmeretal.2017),以逐步更新节点嵌入并预测局部和全局lDDT评分(Mariani等人,2013)。经验评价结果表明,GraphQA实现了与现有的质量评价方法相似的预测精度,降低了所使用的节点/边缘特征的简单性。
在这里,我们使用我们的预训练模型来提取结构嵌入,以进一步增强GraphQA的节点和边缘特征。具体来说,我们将需要评估的结构诱饵输入我们的训练前模型(没有随机扰动),并得到得到的结构嵌入GS。由于特征图中的每个空间位置都对应于一对残差,因此我们通过在相应的位置上选择特征向量来增强边缘特征。类似地,可以通过连接结构嵌入GS的一维池化结果来增强节点特征。然后,GraphQA模型将这些增强的节点和边缘特征作为局部和全局lDDT预测的输入。图2的上部描述了整体的工作流程。
蛋白质间相互作用位点预测
蛋白蛋白相互作用模型预测了两种或多种蛋白质之间的物理接触,这在各种生物过程中起着至关重要的作用(DeLasRivas和丰塔尼罗,2010b;Li等人,2019年)。为了更好地理解不同的蛋白质之间是如何相互作用的,第一步是确定每个蛋白质中的哪些氨基酸残基实际上参与了相互作用。在形式上,我们遵循(Zengetal.2020),如果一个氨基酸在蛋白质结合前后的绝对溶剂可及性小于![]()
,则将其定义为PPI位点。因此,PPI站点预测任务可以看作是一个预残差二值分类问题。在(Zengetal.2020)中,作者提出了DeepPPISP作为一个端到端框架,它集成了局部上下文和全局序列特征,用于PPI位点预测。具体地说,从以每个氨基酸残基为中心的固定大小的滑动窗口中提取局部特征,以捕获局部模式,而通过一维卷积网络提取全局特征。然后,将局部特征和全局特征连接起来,并被后续的分类子网络用于每残差分类。
类似地,我们的预训练模型可以作为一个即插即用模块,以增强DeepPPISP模型中使用的本地和全局特性。
对于PPI位点预测中使用的每个训练样本,我们使用预训练模型对蛋白质结构进行编码,以计算相应的结构嵌入。通过在结构嵌入上应用二维池化,获得了附加的全局特征。对于局部特征,每个残基结构嵌入可以计算为全尺寸结构嵌入的一维池化结果。然后,这些嵌入由相同的滑动窗口分组,以生成额外的局部上下文特征来描述每个氨基酸残基。通过连接所有原始/附加的局部和全局特征,DeepPPISP模型可以通过一个增强的特征集进行训练,用于PPI站点预测。
五、总结
最后,我们演示了如何利用训练前的模型在不同的层次上产生结构嵌入。只要下游任务依赖于蛋白质的基于结构的特征,那么包含我们的结构嵌入来进一步增强其特征表示应该总是有益的。潜在的应用方案包括蛋白质折叠分类(Chenetal.2016)和基于结构的药物设计(Batool,Ahmad和Choi2019)。
六、Experiments
6.1 下游任务的预训练模型
数据集
对于训练前模型,我们从RCSB-PDB数据库(发布于2021年1月1日)(Bermanetal.2000)中获得天然蛋白质结构,其中包括超过17万个未标记的蛋白质三级结构。rcsbpdb数据库有些冗余,其中相同或高度相似的氨基酸序列可能对应于多个蛋白质结构。因此,我们采用官方的序列聚类结果,BC-30和BC-100,分别过滤出序列一致性至少为30%或100%的冗余序列。BC-100数据集去除下游任务中有效和测试数据的重叠蛋白后,包含73585个蛋白质,其中58868个作为训练集,7357个作为验证集,其余为测试集。BC-30数据集由29,242个蛋白质组成。其中,23,394个蛋白质被用作训练集,2,923个蛋白质作为验证集,2,925个蛋白质被用于测试。
对于蛋白质QA预测任务,我们使用GraphQA发布的数据集(Baldassarreetal.2021)。CASP9-CASP12数据集包含85k个诱饵,这些诱饵被随机分成一个训练集(˜270个目标)和一个验证集(˜50个目标)。CASP13数据集在测试集中包含˜14k诱饵(˜72个目标)。
对于PPI位点预测任务,我们使用了来自DeepPPISP(Zeng等,2020年)的处理数据,即186个蛋白中的Dset186个,72个蛋白中的Dset72个(村上和小铃木,2010年)和164个蛋白中的PDBset164个(Singh等,2014年)。DeepPPISP删除了两个蛋白质,因为它们没有相关的蛋白质DSSP文件(Kabsch和Sander1983),这是该方法中使用的输入特性之一。DeepPPISP将三个数据集集成到一个融合的数据集上,以确保训练集和测试集来自相同的分布。我们从(Zengetal.2020)下载训练、验证和测试数据列表。训练集中有300个蛋白质,独立验证集中有50个蛋白质,测试集中有70个蛋白质。
Input features
除了第3节中描述的距离矩阵外,我们还将蛋白质特异性信息编码为评分网络的输入特征,其中包括蛋白质序列单热特征和位置编码(Vaswanietal.2017)。有关特性和编码的详细信息,请见附录C。
网络架构和学习超参数
我们的预训练评分网络采用了全卷积神经网络架构,它由32个具有扩张卷积的剩余块组成。为了减少计算开销,我们对每个残余单元应用了瓶颈机制(Heetal.2016)。我们还使用条件批处理归一化(Song和Ermon2019)来考虑随机噪声的标准偏差水平。隐藏层的通道数k被设置为64。我们还在附录e中报告了不同通道数的结果。我们使用32的批量大小进行训练和验证,并随机裁剪大小为32的输入特征图以进行数据增强。位置编码的维数被设置为dmodel=24。我们为K=32水平构造了随机噪声的标准差,其范围为0.01到10.0。当σ1=10.0时,构象空间可以得到充分的探索,而σK=0.01表明对原生结构引入了微小的扰动。
对于优化,我们应用一个恒定的学习率为0.0001,并使用Adam(Kingma和Ba2014)作为我们的训练前模型的优化器。经过50个epoch的训练后,我们根据验证损失选择最优检查点,然后将其用于即将到来的结构嵌入(GS)生成。
七、Results
表1显示了CASP13数据集对蛋白质QA下游监督任务的比较性能。除了通过运行有和没有我们的预训练模型的实验来评估我们的方法的有效性外,我们还比较了基于蛋白质序列的嵌入的性能。我们使用GraphQA作为基线模型,我们遵循(Raoetal.2019)来生成TAPE的基于序列的嵌入。表1-GDTTS显示了全球质量预测w.r.t.的各种评价指标的结果GDT TS.对于RMSE和FRL5,越低越好;对于R、Rtarget和z,越高越好。结果表明,通过我们的预训练模型生成的嵌入,GraphQA比其他所有方法都更有能力,包括使用原始特征和在对诱饵的总体质量进行排序时添加基于序列的嵌入。有关QA任务评估指标的更多细节,请参见附录C。
本地质量预测的性能w.r.t.CAD和LDDT评分,越高越好。正如所观察到的,我们的预培训方法进一步提高了地方层面的绩效表明我们在当地的嵌入式产品质量很高(残基)水平,以及区分正确预测的蛋白质链部分的能力。因此,我们的预训练模型提取的嵌入可以使预测网络捕获更复杂的信息和残差之间的长期依赖性。请注意,在本地分数上添加序列嵌入的结果比基线的结果要差。一个可能的原因是本地QA任务更依赖于残留间(边缘)信息,而TAPE不包含这些信息。此外,添加大量的维度节点特征(768个维度的磁带)使原始网络更难训练。我们根据GraphQA(Baldassarreetal.2021)中的实验设置精确地实现了实验,包括数据分割、网络超参数和训练策略。蛋白质QA下游任务的其他结果见附录D。
表2显示了有和没有由我们的预训练模型生成的嵌入的DeepPPISP模型训练的结果,我们也引入了TAPE嵌入进行比较。由于DeePPPISP不能为数据加载提供种子,因此我们重复实验5次,得到均值和标准差,以消除随机性,并验证其鲁棒性。虽然我们的方法的召回率低于基线的性能,但所有其他评估指标的分数都是最高的。值得注意的是,PPI站点预测训练问题是不平衡的,因此下游任务通常更集中于MCC和Fmaste的性能(Zengetal.2016),DeepPPISP使用F-meatse来选择最佳验证模型。与QA任务相比,PPI任务中PPI任务对序列信息和结构信息的依赖性相对平衡。
磁带比只利用原始特征的基线模型表现更好的方法是合理的。此外,我们的结构嵌入能够通过探索结构信息来获得更好的性能。此外,我们还在一个较小的数据集上进行了预训练实验,即BC-30过滤数据集,以验证所提方法的有效性。如表1和表2所示,尽管训练前所涉及的数据是精简的,但它在下游任务上始终表现良好。结果表明,即使在较小的数据集上进行预训练,我们的模型仍然可以为下游任务提供高质量的局部和全局嵌入。为了证明我们的预训练模型的鲁棒性,我们还对不同隐藏大小的分数网络进行了实验。由于空间限制,下游任务的相应结果延迟到附录E。结果表明,在使用较小的数据集进行预训练时,利用预训练的结构嵌入方法,在下游任务中仍能取得良好的性能。
八、Conclusion
在这项工作中,我们提出了一个自我监督的蛋白质结构的预训练模型。据我们所知,这是第一次尝试构建和评估蛋白质三维结构上的自监督学习。此外,我们的方法可以很容易地应用于各种下游模型。经验表明,我们的预训练模型可以为下游任务生成高质量的结构嵌入。最近的预训练策略主要集中在蛋白质序列数据集上,因为它更容易获得和包含大量的数据。然而,即使用于预训练蛋白质三维结构的数据集也没有蛋白质序列数据集大,我们认为该三维结构比序列包含更多的信息。为了充分利用现有的蛋白质数据,我们的下一步行动是将三维结构预训练策略与基于序列的预训练方法相结合,以获取足够的蛋白质信息。