【实验技术笔记】RNA 抽提 + 反转录PCR + PCR引物设计 + RT-qPCR
文章目录
- 1. RNA 抽提
- 2. 反转录 PCR (rtPCR)
- 3. PCR 引物设计
- 4. RT-qPCR
1. RNA 抽提
TRIzol 作用原理

● TRIzol 法的优点:
- 快速
- 能抽提多种属总 RNA
- 对少量的组织和细胞以及大量的组织和细胞都能有效分离
- 能抽提不同分子量大小的多种 RNA
- 能够避免 DNA 和蛋白的污染
- 抽提出来的 RNA 适用于多种实验
RNA 抽提前的准备
● 试剂耗材准备:

● 样品准备:

RNA 抽提流程
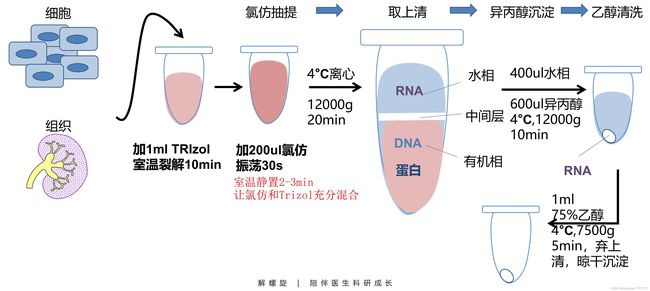
● RNA 抽提操作
- 氯仿抽提:
◆ 每 1mlTRIzol中加 200ul氯仿,用手大力摇管 15s,室温孵育 2-3min;(使氯仿和 TRIzol 充分混匀,不能用振荡器,否则容易使 DNA 的亲水基团与水相接触,造成 DNA 污染)
◆ 4°C 离心 12000g × 15min。(离心后,DNA、蛋白质混合物分离为下层红色的有机相、中间相以及上层无色水相,RNA 存在于水相。) - 取水相: 转移 400ul 水相到新管。(转移水相时,枪尖随着液面下移而下移,避免扰乱白色分层,慢慢吸取,避免吸到中间相和下层有机相,不要贪心吸太多,吸大约 400ul 就好。)
- 异丙醇沉淀:
◆ 加入 600ul异丙醇上下颠倒混匀 10 次,室温孵育 10min(使核酸沉淀下来,分离盐类杂质),或者 4℃或-80℃孵育 1-2h;(低温沉淀能提高核酸沉淀效率,当核酸浓度很低时用低温沉淀效果明显;如果核酸样品比较脏,会大大增加杂质被同步沉淀的几率,不建议使用低温沉淀以及沉淀时间过长)
◆ 4°C 离心 12000g × 10min。(往往离心前 RNA 不可见,离心后在管侧面和底部形成白色沉淀。) - 乙醇清洗: 弃上清,轻轻加入 1ml
75%乙醇洗涤一次,4°C 离心 7500g × 5min。(加入 75%乙醇时让 RNA 沉淀轻轻漂起来,或者保持原位,因为有时候 RNA 太少,吹散了再离心下来时 RNA 碎片不一定能聚集到一起形成肉眼可见的沉淀,导致后面的“盲操作”。) - 晾干沉淀后溶解: 尽量吸除上清,室温干燥 5-10min,以
DEPC treated 水溶解,-80°C 保存。(密切注意 RNA 沉淀的干燥程度,不要完全干燥,这将大大降低其溶解度。部分溶解的 RNA 其 A260/280 比值会偏低。最佳干燥度为 RNA 沉淀微微湿润呈啫喱状。)
RNA 质量检测
1、分光光度计测定:Nanodrop 2000、Agilent 2100
- RNA 在 260nm 处有最大吸收峰,通过测定样品的 OD260、OD280,可以估计样品的含量和纯度。OD260 为 1 的溶液含有 40ug/ml。
- 纯的 RNA 其 OD260/OD280 应为 2.0,如果比值偏小,则有机溶剂或蛋白污染较严重。如果比值偏大,则 RNA 可能已发生降解。
- 以吸光度值估计 RNA 含量和纯度并不十分可靠。需要结合其他方法综合判断。
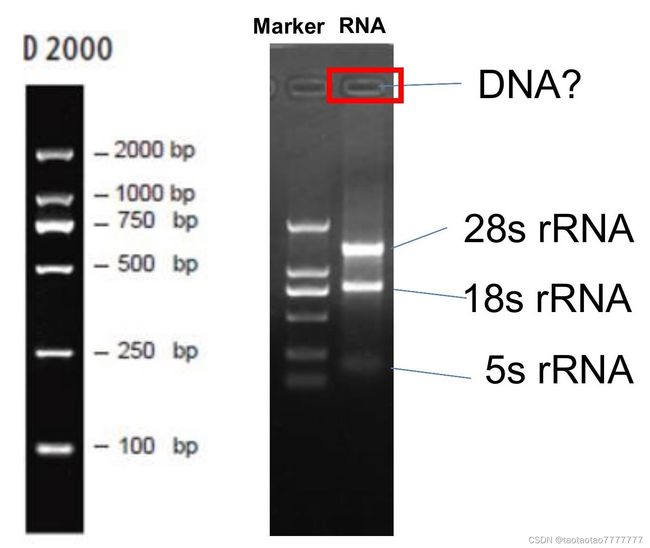
2、凝胶电泳法:
-
配制
1.2% 琼脂糖凝胶。将样品加上RNA loading buffer后加入上样孔电泳。电泳液用新鲜的1×TAE即可。
◆ RNA loading buffer 配方:
-
紫外下拍照可以看见 3 条带,从上往下依次是 28s RNA、18s RNA 和 5sRNA。其中
28sRNA (5kp) 应为18sRNA 亮度的 2 倍且没有涂抹状条带,这样的 RNA 质量是过关的。 -
吸光度测定法来不能区分 DNA 和 RNA,而电泳法如果看到上样孔内亮亮的,那通常为污染的 DNA。如果 DNA 会干扰后续实验,可以用
DNase消化去除。 -
可以通过与 marker 比较估计 RNA 含量。(marker 通常会告诉上多少量的 marker,最亮条带相当于多少量的 DNA,例如 DNA marker 最亮条带是 1ug,上样量约 1.5ug, 因此将 RNA 条带亮度与 marker 对比,可大概得知 RNA 的量。)
2. 反转录 PCR (rtPCR)
- rtPCR:检测 RNA。
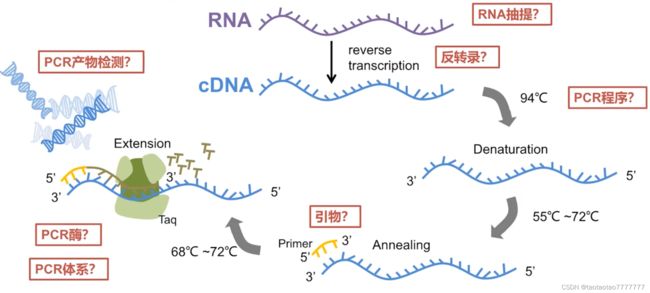
01 反转录
-
反转录实验原理:

-
根据不同情况选择不同的反转录引物:
(1)RNA 有 polyA 尾:用Oligo(dT)作为反转录引物,要求 RNA 质量好,否则 poly(A) 尾可能降解,造成全长 cDNA 合成量大大减少。
(2)RNA 没有 polyA 尾:如原核生物 RNA、真核生物的 rRNA、tRNA,用随机引物 (Random primer) 作为逆转录引物。(几乎所有 RNA 都可以用随机引物来反转)
(3)目的序列已知时,用与模板序列特异性互补的引物作为反转录引物。
★ 推荐试剂盒:TOYOBO ReverTraAce -α- (FSK-101 100 reactions)
-
试剂盒中的反转录酶经过突变改造:
◆ 反转录酶主要功能结构域:polymerase 结构域(负责 cDNA 合成)、RNase H 结构域(降解 RNA)。因此,野生型的反转录酶转录出的产物是单链 cDNA。
◆ 突变的反转录酶失去了 RNase H 的功能,使得 RNA 不被降解,得到的产物是单链 cDNA 和 RNA 的杂合链。 -
反应体系配制及 PCR 条件设置:

02 PCR 扩增
★ PCR 酶和 PCR 体系
-
推荐试剂 1:TaKaRa Premix TaqTM (TaKaRa TaqTM Version 2.0 plus dye)(RR901A 性能普通,适合目的基因好扩的)

◆ 2×mixture,包含酶、buffer、dNTP、Mg2+,使用方便,配制体系前充分混匀。
◆ 已含有染料,跑完 PCR 后不需要再加 loading buffer。
◆ Taq 酶冷启动:在冰上配制反应体系。 -
推荐试剂 2:TaKaRa Ex Taq DNA Polymerase (RR001Q) 冷启动
-
推荐试剂 3:TaKaRa Ex Taq Hot Start Version (RR006Q) 热启动,特异性好,便宜。
★ PCR 程序设置

- 退火温度:55-60℃,可以直接用引物 Tm 值-5℃。
- 延伸时间:
一般 Taq 酶的速度: 1 min/kb( 2kb 以内);1 min/500bp(2kb 以上)
一般控制扩增产物在 300-400bp, 例如,扩增 400bp 的 400*60/1000 =24s。 - 终止延伸时间:补齐所有没有延伸完成的 DNA 链,提高完整双链 DNA 的比例,一般设置 5min 足够。
03 PCR 产物检测
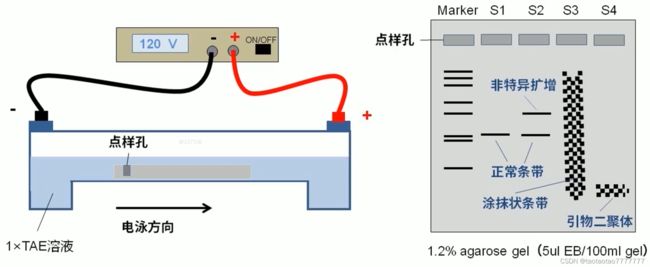
-
出现涂抹状条带的原因:
① Mg2+加多了,导致酶活性异常。商品化的试剂一般不存在这个问题。
② 电泳缓冲液不新鲜,需要更换了。 -
出现引物二聚体的原因:
引物设计不好:引物设计时,应保证产物大于与引物二聚体,保证电泳结果可以明显区分出来;
PCR 条件设置不好,导致引物不能很好的结合到模板。 -
50×TAE 配方:

RT-PCR 出问题的原因
1、无扩增产物:
-
可能原因 1:引物问题(包括:引物设计较差,引物合成较差,引物浓度太低)。
解决方法:设计引物的时候,避免在引物 3’端含有互补序列;避免可以形成内部发卡结构的序列;设计 Tm 类似的引物。针对引物合成的问题,用普通电泳法,看引物的设计和合成质量,是否有目的条带出现。最佳引物浓度介于 0.1μM 到 0.5μM 之间。 -
可能原因 2:探针问题。
解决办法:用 blast 对探针序列进行比对,设计符合要求的探针。 -
可能原因 3:模板问题(包括:模板质量不好,模板浓度太低)。
解决方法:提高模板质量。如果怀疑模板被 DMSO 等抑制剂污染了,坚决采用乙醇沉淀;使用 104copy 的靶序列,然后在 25 到 30 个循环中获得信号。 -
可能原因 4:不明物干扰。
解决办法:做 RT-PCR 必须非常小心翼翼。对 RT-PCR 的任何实验样品或试剂,均应采用“三无”离心管(即无菌无酶无热源)进行试剂的分装和使用。 -
可能原因 5:扩增酶问题。
解决办法:不要省钱。买好的扩增酶。分子生物学永远是一分价钱一分货。推荐:选择 hot start Taq 酶进行扩增,可提高灵敏度,增加产量,降低干扰因素。
2、非特异信号:
-
可能原因 1:引物问题(包括:引物二聚体太多)。
解决方法:检查引物设计环节和合成环节,必要时重新设计和合成引物。 -
可能原因 2:扩增酶问题。
解决办法:选择hot start Taq 酶进行扩增,可提高灵敏度,增加特异性。 -
可能原因 3:检测温度太低。
解决办法:在更高的温度下检测荧光信号。这个时候,引物二聚体已经解链。
3、灵敏度低:
-
可能原因 1:RNA 问题(包括:RNA 被损害和降解;初始模板 RNA 不充分)。
解决方法:如果完全损害或者降解的话,更换 RNA;增加模板 RNA 的浓度;使用 10ng-1μg 的总 RNA。 -
可能原因 2:仪器故障。
解决方法:检查扩增仪的温度;检查荧光仪器温度和信号。如果发现故障,找厂家来修。 -
可能原因 3:PCR 扩增无效。
解决方法:反转录的抑制剂有很多,包括 SDS、EDTA、胍盐、甲酰胺、磷酸钠和亚精胺等等。一定要注意,不要被这些抑制剂污染了。
3. PCR 引物设计
引物设计流程
- 找到目的基因(各转录本)CDS 序列的保守区域
- 用软件设计引物
- 用软件评价引物
- Blast 引物特异性
引物设计操作
- 引物设计软件:Oligo
参考:常用生物学软件的安装与应用(六) —Oligo7 - 序列比对软件:DNAMAN
参考:常用生物学软件的安装与应用(二) —DNAMAN - 普通 PCR 引物设计的优先选择顺序为 Primer-BLAST/Primer3 > Primer6 > Oligo7 > SnapGene >DNAMAN,分子克隆引物设计优先使用 SnapGene。
3 款简单实用的在线 PCR 引物设计软件
Primer3web version 4.1.0
Primer3web 在线引物设计–2 分钟学会步骤极简的引物设计! - 步骤:
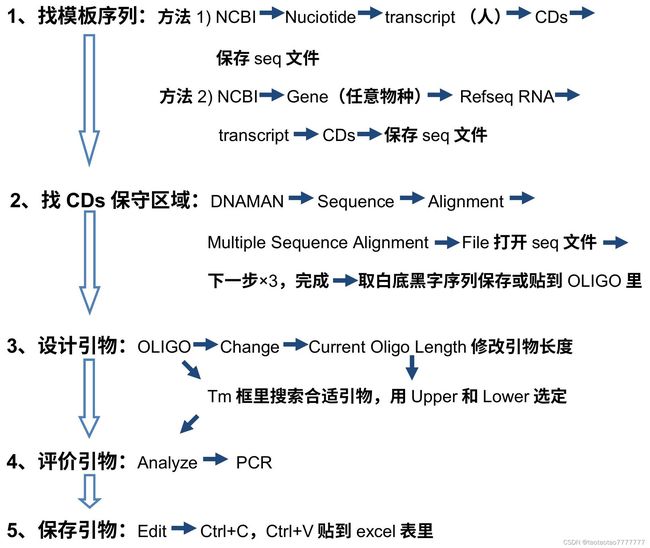
-
找模板序列:以 TP53 为例,只看确定的转录本,不看预测的。
方法 1):NCBI-Nucleotide中搜索目的基因的transcript(人)→ CDS → 保存.seq文件
方法 2):NCBI-Gene(任意物种)→Refseq RNA→transcript→ CDS → 保存.seq文件
 2. 找 CDS 保守区域:
2. 找 CDS 保守区域: DNAMAN→ Sequence → Alignment → Multiple Sequence Alignment → File 打开 seq 文件 → 下一步×3,完成 → 取白字黑底序列(所有转录本共有的保守序列)保存或贴到 Oligo 里(选中序列 → Edit → Copy → text)。
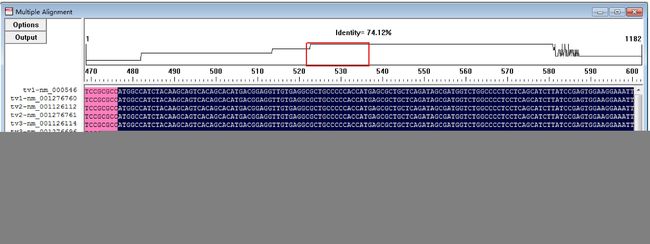
-
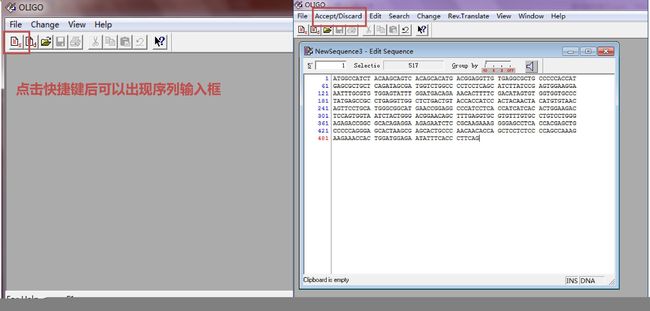
设计引物:
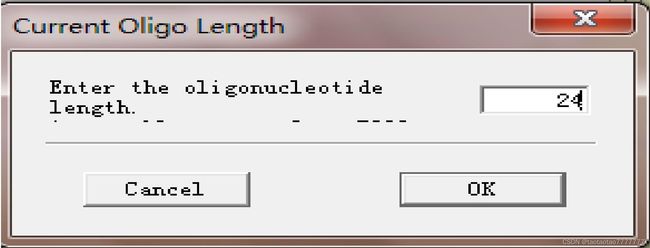
Oligo 6输入序列 →Accept→ 选择 Tm 框Melting Temperature→Change→Current Oligo Length修改引物长度(常用 23-24bp) → Tm 框里搜索合适引物,用Upper和Lower选定。
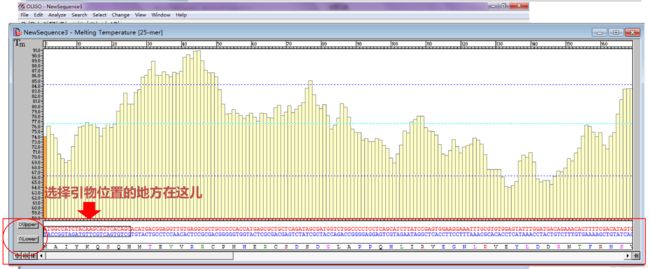
◆ 设置引物长度
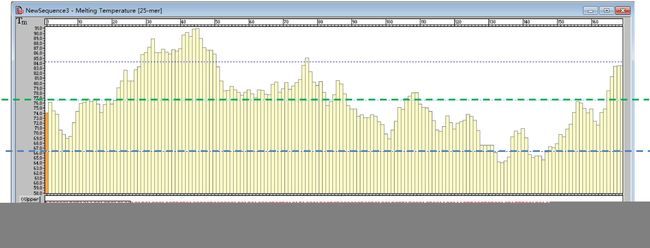
◆ 肉眼搜索引物:
① 平均 TM 值在72℃左右(下面两条虚线之间。Taq 酶在 72℃ 进行 DNA 复制,DNA 解链,引物与 DNA 单链结合)
② 5’端 TM 值高于 3’端(因为合成方向是 5’-3’,即 3’端会攻击 DNA 链使得引物与 DNA 链结合,能量越低越容易发生攻击反应。)
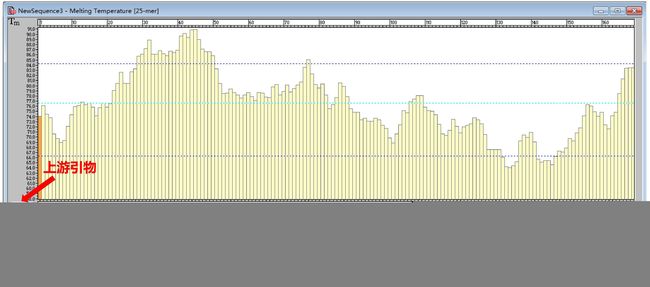
◆ 选择引物位置
-
评价引物:上下游引物的 Tm 值要相近(72℃左右),可以通过修改引物长度(
Change→Current Oligo Length)来调整。
◆ 查看引物参数:Analyze→PCR
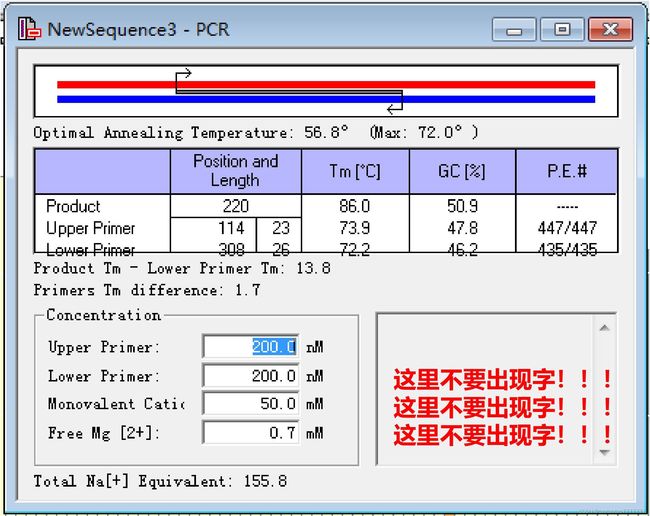
◆ 框框里出现提示信息怎么办?
①3'-end dimer between the primers.引物间有二聚体,那就随意挑一条引物挪动几个碱基;
②Two-temperature PCR-recommened: (68°/94°)有两个可用的温度,那就还是随意挑一条引物挪动;
③Terminal stability of the Lower Primer is too high.下游引物的一端稳定性太高,那就是说这条引物的碱基对应着的柱子高了,把它往柱子低一点的地方挪;
④Terminal stability of the Upper and Lower Primers is too high.上下游引物的末端稳定性都太高,那就把两条都挪往低一点的地方。 -
保存引物:两种方法
(1)Edit→Upper Primer/Lower Primer→ Ctrl+C,Ctrl+V (软件会自动按 5’-3’方向显示引物序列)贴到 excel 表里(填引物订单时)。
(2)File→Save保存成 .seq 文件。

-
BLAST 一下确认引物是否会扩增出其他基因(确保正反向都只能扩出目的基因),不能则可以合成。
参考:3款简单实用的在线PCR引物设计软件 搜索关键词:NCBI blast 引物验证
引物设计要点
- 引物长度:18~30bp。—— Change 改变引物长度
- 引物 GC 含量:40%~60% ——选两条虚线间的碱基
- Tm 值:72 ℃左右,上下游引物的 Tm 保持一致。(选择合适的位置,设置合适的引物长度,3’端 Tm 比 5’端低)。
- 避免扩增模板的二级结构区域 ——选择合适位置的碱基
- 避免引物的二级结构 ——选择合适位置的碱基
- 避免与靶 DNA 的错配 ——尤其注意 3’端
- 也要注意产物长度,rtPCR 的产物长度控制在
300-500bp,如果实在找不到合适引物,标准可适当放宽。
4. RT-qPCR
RT-qPCR
● RT-qPCR:以 cDNA 为模板进行 PCR,在 PCR 扩增过程中,通过收集荧光信号,对 PCR 进程进行实时检测。由于在 PCR 扩增的指数时期,模板的 Ct 值和该模板的起始拷贝数存在线性关系,所以可以定量。
- 探针类:包括
TaqMan 探针和分子信标,利用与靶序列特异杂交的探针来指示扩增产物的增加; - 非探针类:其中包括如
SYBR Green I或者特殊设计的引物(如 LUX Primers) 通过荧光染料来指示产物的增加。
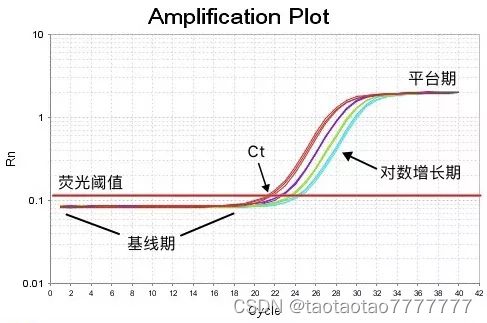
在实时 PCR 扩增过程中,荧光信号被收集,转化为成为扩增和熔解曲线。具体数据就是基线,荧光阈值和 Ct 值。
- 基线(
baseline):一般来讲,第3-15 个循环的荧光值就是基线,是由于测量的偶然误差引起的。 - 阈值(
threshold):阈值一般是基线的标准偏差的 10 倍。在实际操作中也可以手动调节,位于指数期就可以。 - Ct 值(
Ct value):Ct 值就是荧光值达到阈值时的 PCR 循环次数。所以是一个没有单位的参数。与初始模板的量呈反比。一般来说 Ct 值在 30 以下都可以说实验结果是可靠的,30 以上基本可以说明该基因没有扩增,但也不是的,需要辅以溶解曲线加以解释。
● SYBR Green 法原理
参考:SYBR Green I 法原理/优缺点/常见问题解析
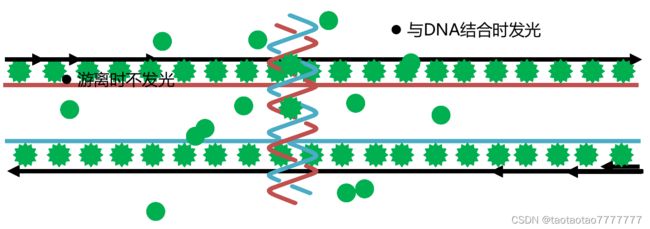
- SYBR Green 是双链 DNA 结合染料,在 PCR 反应体系中,Sybr 荧光染料特异性地掺入 DNA 双链与双链 DNA 结合后,发射荧光信号,而不掺入双链中的 Sybr 染料分子不发射任何荧光信号。从而保证与 PCR 产物的增加完全同步。
- 优点:
1、对 DNA 模板没有选择性;
2、使用方便,无需复杂的探针设计
3、成本较低使用方便 - 缺点:
1、实验容易产生假阳性
2、对引物特异性要求较高
3、灵敏度相对较低
RT-qPCR 操作流程
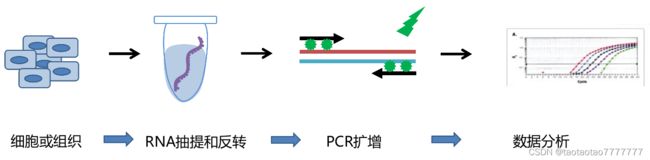
- 推荐试剂:TOYOBO ReverTra Ace® qPCR RT Master Mix with gDNA Remover (可以去除基因组 DNA,只剩 RNA,设计引物时就不用考虑要不要跨外显子)
RT- qPCR 实验操作要点
-
1、引物设计
除了遵循一般的引物设计原则外,需要注意产物长度应控制在 80-150bp。
片段越短扩增效率越高,但若小于 75bp 则无法与潜在的引物二聚体区分,从而给判断引物优劣带来障碍。
推荐:PrimerBank、Oligo6、paper
参考:3 款简单实用的在线 PCR 引物设计软件 -
2、Mix 配制
推荐试剂:DBI Bioscience Bestar® Sybr Green qPCR Master Mix (热启动)

◆ 根据所用 MasterMix,模板和引物的不同进行优化,达到一个最佳反应体系。注意是否添加 ROX(校正染料)。
◆ MasterMix 不要反复冻融,如果经常使用,最好溶解后放在 4 度。
◆ 冰上操作。
◆ 每管或每孔都要换新枪头!不要连续用同一个枪头加样!减少加样误差!
◆ 所有成分加完后,离心去除气泡(以免气泡影响荧光信号收集)。
◆ 每个样品至少 3 个平行孔。ROX(Passive Reference Dye)是一种惰性参比染料,在 qPCR 反应中能被 qPCR 仪检测到。ROX 不参与 qPCR 反应,荧光信号值不会随着 qPCR 扩增而改变。实验过程中很多因素会引起孔间差异:不均匀的照明,孔与孔之间光学因素(液滴、气泡)的因素,反应体系的浓度不同…… 可以用 ROX 作为阳性参比,对其他荧光信号进行归一化校正,以提高数据的较精确度。
Real-time qPCR 数据分析——(1)相对定量
● 相对定量:检测实验组和对照组中一个靶基因的倍数差异。
- 如果对 RNA 模板的数量不能精确定量,或者只需要知道目的基因的表达差异时,可以使用相对定量法。
- 需要进行归一化处理。
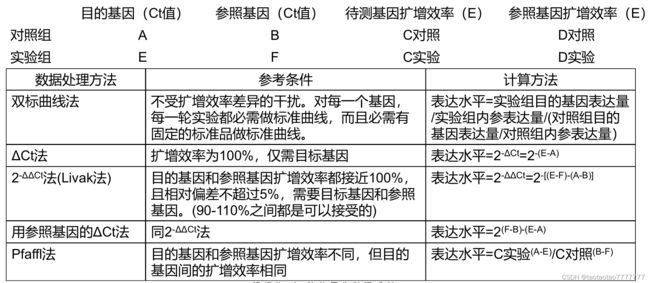
★ 相对定量计算方法:
-
常用的相对定量数据分析方法有双标曲线法,ΔCt 法,2-ΔΔCt 法 (Livak 法),用参照基因的 ΔCt 法和 Pfaffl 法。
-
ΔCt 法:不用内参基因作为标准,实验设计简单,数据分析处理简单。需要准确量化初始材料(如细胞数目或核酸微克数),扩增效率为 100%。计算公式如下:

-
2-ΔΔCt 法:这是最常用的进行相对基因表达分析的方法,得到的结果是实验组中目的基因相对于对照组中目的基因表达的差异倍数。要求目的基因和内参基因的扩增效率都接近 100%,且相对偏差不超过 5%。计算公式如下:
Step1:用内参基因 Ct 值对实验组 (test) 和对照组 (con) 的靶基因 Ct 值归一化:
ΔCt test = 实验组目的基因 Ct 值- 实验组内参基因 Ct 值
ΔCt con = 对照组目的基因 Ct 值- 对照组内参基因 Ct 值
Step2:用对照组的 ΔCt 值归一化实验组的 ΔCt:ΔΔCt = ΔCttest-ΔCtcon
Step3:计算表达水平的差异倍数:Change Fold = 2-ΔΔCt -
用参照基因的 ΔCt 法:这个方法的使用前提与 2-ΔΔCt 法相同。计算方法:

◆ 这种方法得到的对照组表达水平不是 1.0,但如果将得到的两个表达值都除以对照组表达值,则得到:

得到的比值与 2-ΔΔCt 法是相同的,因此 用参照基因的 ΔCt 法是 2-ΔΔCt 的一种变化形式。 -
Pfaffl 法:当目的基因扩增效率 (E target) 和内参基因扩增效率 (E ref) 不同,但每个基因在实验组和对照组扩增效率相同时,可以按下列计算公式确定表达差异。2-ΔΔCt 法是 Pfaffl 法的简单特例。
Real-time qPCR 数据分析——(2)绝对定量
参考:干货|绝对定量之 DNA 拷贝数计算
● 绝对定量:是用一系列已知浓度的标准品制作标准曲线,在相同的条件下目的基因测得的荧光信号量同标准曲线进行比较,从而得到目的基因的量。
- 标准品可以是纯化的质粒 DNA、体外转录的 RNA、体外合成的 ssDNA。
- 检测给定数量的样品中靶基因 mRNA 拷贝数。
- 如果想知道每毫升样品中的某基因的拷贝数时,使用绝对定量。
- 需要校准。
● 如何使用标准曲线进行绝对定量?
在进行绝对定量前,我们首先需要使用已知浓度的模板建立标准曲线,这可以通过梯度稀释已知样品,使用这些稀释的样品作为标准品来构建标准曲线。
- 标准品建立
标准品通常可以使用含有待检测基因的质粒。通过测定质粒浓度可以很方便计算出标准品中基因的拷贝数。计算方法如下:
质粒浓度(ng/ul)= OD260×50×稀释倍数
待测基因分子量 = 碱基数×324
样本拷贝数(copies/ul)= 质粒浓度/待测基因分子量×6×1014 - 标准品和待测样品准备
将计算好拷贝数的含待测基因的质粒做梯度稀释,使用 SYBR Green 建立反应体系。同时取待测样品置于同一实验中进行反应。 - 标准曲线绘制
以标准品的拷贝数为 x 轴,Ct 值为 y 轴绘制标准曲线,可以得到标准曲线公式:y=k*x+b,将未知样品的 Ct 值代入,就能得到相对应的拷贝数。
Real-Time qPCR 常见问题分析
参考:qPCR 常见问题及解决方案
1、无 Ct 值出现
- 检测荧光信号的步骤有误:一般 SybrGreen 法采用 72℃延伸时采集,Taqman 法则一般在退火结束时或延伸结束采集信号。
- 引物或探针降解:可通过 PAGE 电泳检测其完整性。
- 模板量不足:对未知浓度的样品应从系列稀释样本的最高浓度做起。
- 模板降解:避免样品制备中杂质的引入及反复冻融的情况。
2、Ct 值出现过晚(Ct>38)
- 扩增效率低:反应条件不够优化。设计更好的引物或探针;改用三步法进行反应;适当降低退火温度;增加镁离子浓度等。
- PCR 各种反应成分的降解或加样量的不足。
- PCR 产物太长:一般采用 80-150bp 的产物长度。
3、标准曲线线性关系不佳
- 加样存在误差:使得标准品不呈梯度。
- 标准品出现降解:应避免标准品反复冻融,或重新制备并稀释标准品。
- 引物或探针不佳:重新设计更好的引物和探针。
- 模板中存在抑制物,或模板浓度过高。
4、负对照有信号
- 引物设计不够优化:应避免引物二聚体和发夹结构的出现。
- 引物浓度不佳:适当降低引物的浓度,并注意上下游引物的浓度配比。
- 镁离子浓度过高:适当降低镁离子浓度,或选择更合适的 mix 试剂盒。
- 模板有基因组的污染:RNA 提取过程中避免基因组 DNA 的引入,或通过引物设计避免非特异扩增。
5、溶解曲线不止一个主峰
- 引物设计不够优化:应避免引物二聚体和发夹结构的出现。
- 引物浓度不佳:适当降低引物的浓度,并注意上下游引物的浓度配比。
- 镁离子浓度过高:适当降低镁离子浓度,或选择更合适的 mix 试剂盒。
- 模板有基因组的污染:RNA 提取过程中避免基因组 DNA 的引入,或通过引物设计避免非特异扩增。
6、扩增效率低
- 反应试剂中部分成分特别是荧光染料降解。
- 反应条件不够优化:可适当降低退火温度或改为三步扩增法。
- 反应体系中有 PCR 反应抑制物:一般是加入模板时所引入,应先把模板适度稀释,再加入反应体系中,减少抑制物的影响。
7、同一试剂在不同仪器上产生不同的曲线,如何判断?
- 判断标准:扩增效率,灵敏度,特异性
- 如果扩增效率在 90%-110%,都是特异性扩增,都可以把数据用于分析。
8、扩增曲线的异常?比如“S”型曲线?
- 参比染料设定不正确 (MasterMix 不加参比染料时,选 NONE)
- 模板的浓度太高或者降解
- 荧光染料的降解