Gromacs学习记录-漏斗网蜘蛛毒素肽的溶剂化研究
学习 教程GROMACS教程:漏斗网蜘蛛毒素肽的溶剂化研究:Amber99SB-ILDN力场|Jerkwin (coding-pages.com) http://t066v5.coding-pages.com/GMX/GMXtut-0/#%E5%88%86%E5%AD%90%E5%8A%A8%E5%8A%9B%E5%AD%A6%E6%A8%A1%E6%8B%9F
http://t066v5.coding-pages.com/GMX/GMXtut-0/#%E5%88%86%E5%AD%90%E5%8A%A8%E5%8A%9B%E5%AD%A6%E6%A8%A1%E6%8B%9F
完全按教程进行,不同的地方进行了加粗
1.在/opt/work1 进行
下载文件:wget http://www.rcsb.org/pdb/files/1OMB.pdb
修改work1文件夹权限
sudo chmod 777 /opt/work1
gmx pdb2gmx -ignh -ff amber99sb-ildn -f fws.pdb -o fws.gro -p fws.top -water tip3p
有警告。忽略。
得到三个文件

gmx editconf -f fws.gro -o fws-PBC.gro -bt dodecahedron -d 1.2
Read 495 atoms
Volume: 0.001 nm^3, corresponds to roughly 0 electrons
No velocities found
system size : 2.793 1.951 2.502 (nm)
diameter : 3.319 (nm)
center : -0.156 -2.386 -0.412 (nm)
box vectors : 0.100 0.100 0.100 (nm)
box angles : 90.00 90.00 90.00 (degrees)
box volume : 0.00 (nm^3)
shift : 4.445 6.675 2.434 (nm)
new center : 4.289 4.289 2.022 (nm)
new box vectors : 5.719 5.719 5.719 (nm)
new box angles : 60.00 60.00 90.00 (degrees)
new box volume : 132.24 (nm^3)gmx grompp -f em-vac-pme.mdp -c fws-PBC.gro -p fws.top -o em-vac.tpr
没有'em-vac-pme.mdp这个文件
把一个pdb文件内容改成下面的 然后名字改成em-vac-pme.mdp,重新运行
11个warning
; 传递给预处理器的一些定义
define = -DFLEXIBLE ; 使用柔性水模型而非刚性模型, 这样最陡下降法可进一步最小化能量
; 模拟类型, 结束控制, 输出控制参数
integrator = steep ; 指定使用最陡下降法进行能量最小化. 若设为`cg`则使用共轭梯度法
emtol = 500.0 ; 若力的最大值小于此值则认为能量最小化收敛(单位kJ mol^-1^ nm^-1^)
emstep = 0.01 ; 初始步长(nm)
nsteps = 1000 ; 在能量最小化中, 指定最大迭代次数
nstenergy = 1 ; 能量写出频率
energygrps = System ; 要写出的能量组
; 近邻列表, 相互作用计算参数
nstlist = 1 ; 更新近邻列表的频率. 1表示每步都更新
ns_type = grid ; 近邻列表确定方法(simple或grid)
coulombtype = PME ; 计算长程静电的方法. PME为粒子网格Ewald方法, 还可以使用cut-off
rlist = 1.0 ; 短程力近邻列表的截断值
rcoulomb = 1.0 ; 长程库仑力的截断值
vdwtype = cut-off ; 计算范德华作用的方法
rvdw = 1.0 ; 范德华距离截断值
constraints = none ; 设置模型中使用的约束
pbc = xyz ; 3维周期性边界条件gmx grompp -f em-vac-pme.mdp -c fws-PBC.gro -p fws.top -o em-vac.tpr
整合文件
400个error
重新来,估计原来的pdb文件处理错误
gmx pdb2gmx -ignh -ff amber99sb-ildn -f fws.pdb -o fws.gro -p fws.top -water tip3p
sudo gmx editconf -f fws.gro -o fws-PBC.gro -bt dodecahedron -d 1.2
gmx grompp -f em-vac-pme.mdp -c fws-PBC.gro -p fws.top -o em-vac.tpr

忽略这个warning
sudo gmx grompp -f em-vac-pme.mdp -c fws-PBC.gro -p fws.top -o em-vac.tpr -maxwarn 5
gmx mdrun -v -deffnm em-vac
目前得到的文件

em-sol-pme.mdp文件内容
define = -DFLEXIBLE
integrator = steep
emtol = 250.0
nsteps = 5000
nstenergy = 1
energygrps = System
nstlist = 1
ns_type = grid
coulombtype = PME
rlist = 1.0
rcoulomb = 1.0
rvdw = 1.0
constraints = none
pbc = xyzgmx grompp -f em-sol-pme.mdp -c fws-b4ion.gro -p fws.top -o ion.tpr
gmx genion -s ion.tpr -o fws-b4em.gro -neutral -conc 0.15 -p fws.top
gmx grompp -f em-sol-pme.mdp -c fws-b4em.gro -p fws.top -o em-sol.tpr
报错 缺少位置限制文件
Fatal error:
Cannot find position restraint file restraint.gro (option -r).
From GROMACS-2018, you need to specify the position restraint coordinate files
explicitly to avoid mistakes, although you can still use the same file as you
specify for the -c option.第六步: 位置限制性预平衡模拟
NVT模拟
sudo gmx grompp -f nvt-pr-md.mdp -c em-sol.gro -p fws.top -o nvt-pr.tpr -r em-sol.gro
正常运行
gmx mdrun -deffnm nvt-pr
Using 1 MPI thread
Using 8 OpenMP threads
starting mdrun 'OMEGA-AGA-IVB in water'
50000 steps, 100.0 ps.
Writing final coordinates.
Core t (s) Wall t (s) (%)
Time: 2553.955 319.244 800.0
(ns/day) (hour/ns)
Performance: 27.064 0.887写npt-pr-md.mdp文件
define = -DPOSRES
integrator = md
dt = 0.002
nsteps = 50000
nstxout = 500
nstvout = 500
nstfout = 500
nstenergy = 500
nstlog = 500
energygrps = Protein Non-Protein
nstlist = 5
ns-type = Grid
pbc = xyz
rlist = 1.0
coulombtype = PME
pme_order = 4
fourierspacing = 0.16
rcoulomb = 1.0
vdw-type = Cut-off
rvdw = 1.0
Tcoupl = v-rescale
tc-grps = Protein Non-Protein
tau_t = 0.1 0.1
ref_t = 300 300
DispCorr = EnerPres
; 压力耦合
Pcoupl = Parrinello-Rahman ; Parrinello-Rahman控压器.
Pcoupltype = Isotropic ; isotropic 指盒子可以平均地向各个方向(x, y,z)膨胀或压缩以维持一定的压力. 进行膜模拟时需要用semiisotropic.
tau_p = 2.0 ; 压力耦合的时间常数(单位ps).
compressibility = 4.5e-5 ; 溶剂的压缩系数(4.5e-5为水在300 K和标准大气压下的压缩系数).
ref_p = 1.0 ; 压力耦合的参考压力(单位bar, 1大气压约为0.983 bar).
refcoord_scaling = com
gen_vel = no ; 不产生速度
constraints = all-bonds
continuation = yes
constraint_algorithm = lincs
lincs_iter = 1
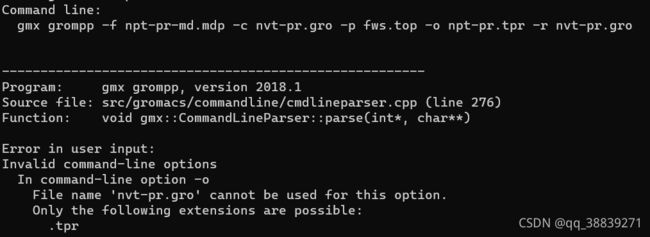
lincs_order = 4sudo gmx grompp -f npt-pr-md.mdp -c nvt-pr.gro -p fws.top -o npt-pr.tpr -r nvt-pr.gro
报错
Error in user input:
Invalid command-line options
In command-line option -o
File name 'nvt-pr.gro' cannot be used for this
option.
Only the following extensions are possible:
.tpr
查手册:When using position restraints, a file with restraint coordinates must be supplied with -r (can be the same file as supplied for -c). For free energy calculations, separate reference coordinates for the B topology can be supplied with -rb, otherwise they will be equal to those of the A topology.(gmx grompp — GROMACS 2018.8 documentation)
-r可以和-c选择一样的文件
NPT模拟
gmx mdrun -deffnm npt-pr

修改mdp文件:在UE里修改并转化为unix文件 再保存
sudo gmx grompp -f npt-pr-md.mdp -c nvt-pr.gro -p fws.top -o npt-pr.tpr -r nvt-pr.gro
nohup gmx mdrun -deffnm npt-nopr &
2021年11月5日21:48:12-->
运行了一晚上(垃圾笔记本)没结果。
2021年11月6日11:28:39
接着NPT模拟
sudo gmx grompp -f npt-pr-md.mdp -c nvt-pr.gro -p fws.top -o npt-pr.tpr -r nvt-pr.gro
残忍报错。。。。
gmx grompp -f npt-pr-md.mdp -c nvt-pr.gro -p fws.top -o npt-pr.tpr -r nvt-pr.gro
这个没报错
gmx mdrun -deffnm npt-pr

cpu爆炸操作

终于到最后模拟了吧
第七步: 成品模拟
sudo gmx grompp -f npt-nopr-md.mdp -c npt-pr.gro -p fws.top -o npt-nopr.tpr -r npt-pr.gro

nohup gmx mdrun -deffnm npt-nopr &
2021年11月6日11:55:58

32核云计算 两分钟跑完,笔记本电脑还在毫无动静。
云计算结果放在work1_result
cd pemission denied
改权限,直接改 opt文件夹权限没有用
sudo chmod 777 /opt/work1_result
使用trjconv命令压缩trr轨迹文件
sudo gmx trjconv -f npt-nopr.trr -s npt-nopr.tpr -o npt-nopr.xtc -pbc nojump -ur compact -center
提示选择时, 两次都选择0.
分析:
gmx ngmx -f npt-nopr.trr -s npt-nopr.tpr
打不开trr文件,没有ngmx
下载VMD,百度搜索下载、
VMD展示trr文件 参考文章:用vmd将gromacs轨迹转换为动画 - 简书 (jianshu.com)
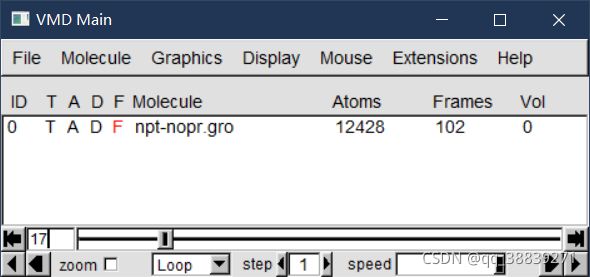
拖动 下面的轴,图像会发生变化

sudo gmx energy -f npt-nopr.edr -o enrg-npt.xvg
得到xvg数据文件,用记事本打开,把数据复制到excel分列以后可以作图。
后面按文章进行。
读取文件加sudo。