TorchProtein教程--基于序列的蛋白质特性预测(3)
TorchProtein教程–基于序列的蛋白质特性预测(3)
本教程来自唐建团队的开源框架torchprotein

目录
- torchprotein安装
- 蛋白质数据结构
- 基于序列的蛋白质特性预测
- 基于结构的蛋白质特性预测
- 预训练的蛋白质结构表示
蛋白质序列表示模型
TorchProtein 定义了多种基于序列的模型来学习蛋白质序列表示。在本教程中,我们使用双层 1D CNN 作为所有考虑任务的蛋白质序列表示模型。首先,让我们通过models.ProteinCNN模块来定义这样一个模型。
from torchdrug import models
model = models.ProteinCNN(input_dim=21,
hidden_dims=[1024, 1024],
kernel_size=5, padding=2, readout="max")
任务 1:蛋白质属性预测
我们想要解决的第一类任务是预测蛋白质属性。我们以β-内酰胺酶活性预测任务为例,旨在预测TEM-1β-内酰胺酶蛋白的突变效应。
在定义数据集之前,我们需要首先定义要在蛋白质上执行的转换。我们考虑两种转换。(1)为了降低基于序列的模型的存储成本,通常的做法是截断过长的蛋白质序列。在TorchProtein中,我们可以通过max length参数指定蛋白质的最大长度,并通过随机参数指定是从随机残基截断还是从第一个残基截断来定义蛋白质截断转换。(2)此外,由于我们想使用残差特征作为基于序列的模型的节点特征,我们还需要为蛋白质定义一个视图变更转换。在数据集构造过程中,可以将两个转换的组合作为参数传递。
from torchdrug import transforms
truncate_transform = transforms.TruncateProtein(max_length=200, random=False)
protein_view_transform = transforms.ProteinView(view="residue")
transform = transforms.Compose([truncate_transform, protein_view_transform])
然后我们通过构建数据集datasets.BetaLactamase,其中会自动下载数据集文件。在这个数据集中,每个样本的标签都是一个实数,表示蛋白质的适应度值。通过打开residue_only选项,TorchProtein 将用于data.Protein.from_sequence_fast加载蛋白质,因此速度更快。我们可以通过该方法得到预先指定的训练、验证和测试拆分split()。
from torchdrug import datasets
dataset = datasets.BetaLactamase("~/protein-datasets/", atom_feature=None, bond_feature=None, residue_feature="default", transform=transform)
train_set, valid_set, test_set = dataset.split()
print("The label of first sample: ", dataset[0][dataset.target_fields[0]])
print("train samples: %d, valid samples: %d, test samples: %d" % (len(train_set), len(valid_set), len(test_set)))
数据结构:
- “sequences”
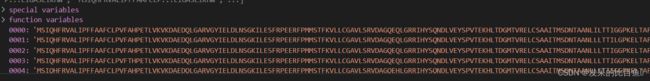
- “targets”:

为了执行 β-内酰胺酶活性预测,我们将 CNN 编码器包装到tasks.PropertyPrediction模块中,该模块将任务特定的 MLP 预测头附加到 CNN 上。
from torchdrug import tasks
task = tasks.PropertyPrediction(model, task=dataset.tasks,
criterion="mse", metric=("mae", "rmse", "spearmanr"),
normalization=False, num_mlp_layer=2)
现在我们可以训练我们的模型了。我们为我们的模型设置了一个优化器,并将所有内容放在一个引擎实例中。在此任务上训练我们的模型 10 个时期大约需要 2 分钟。我们最终评估了它在验证集上的表现。
import torch
from torchdrug import core
optimizer = torch.optim.Adam(task.parameters(), lr=1e-4)
solver = core.Engine(task, train_set, valid_set, test_set, optimizer,
gpus=[0], batch_size=64)
solver.train(num_epoch=10)
solver.evaluate("valid")
评估结果可能类似于以下内容。
任务 2:残基属性预测
我们考虑的第二类任务是预测残差属性。我们以二级结构预测任务为例。
我们首先通过datasets.SecondaryStructure构建数据集,其中我们使用cb513测试集。对于残基级任务,我们通常保留整个蛋白质序列,所以我们只在 ProteinView 这里使用变换。该target字段表示每个残基的二级结构(线圈、链或螺旋),该mask字段表示每个二级结构标签是否有效。两个字段都具有相同长度的蛋白质序列。
dataset = datasets.SecondaryStructure("~/protein-datasets/", atom_feature=None, bond_feature=None, residue_feature="default", transform=protein_view_transform)
train_set, valid_set, test_set = dataset.split(["train", "valid", "cb513"])
print("SS3 label: ", dataset[0]["graph"].target[:10])
print("Valid mask: ", dataset[0]["graph"].mask[:10])
print("train samples: %d, valid samples: %d, test samples: %d" % (len(train_set), len(valid_set), len(test_set)))
数据结构:
- “sequences”
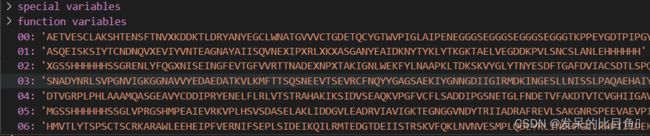
- “targets” : 字段: ‘valid_mask’和’ss3’

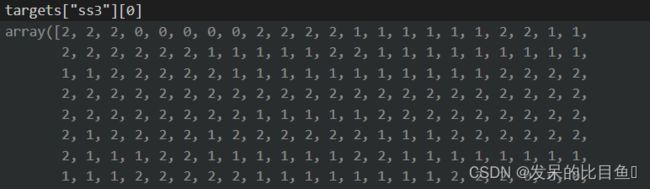
为了执行二级结构预测,我们将 CNN 编码器包装到tasks.NodePropertyPrediction模块中,该模块在 CNN 上附加了一个任务特定的 MLP 预测头。
task = tasks.NodePropertyPrediction(model, criterion="ce",
metric=("micro_acc", "macro_acc"),
num_mlp_layer=2, num_class=3)
构建目标数据集
def target(self, batch):
graph = batch["graph"]
valid_mask = graph.mask
residue_position = graph.residue_position
range = torch.arange(graph.num_residue, device=self.device)
node_in, node_out = functional.variadic_meshgrid(range, graph.num_residues, range, graph.num_residues)
dist = (residue_position[node_in] - residue_position[node_out]).norm(p=2, dim=-1)
label = (dist < self.threshold).float()
mask = valid_mask[node_in] & valid_mask[node_out] & ((node_in - node_out).abs() >= self.gap)
return {
"label": label,
"mask": mask,
"size": graph.num_residues ** 2
}
我们训练模型 5 个epochs,大约需要 5 分钟,最后在验证集上对其进行评估
optimizer = torch.optim.Adam(task.parameters(), lr=1e-4)
solver = core.Engine(task, train_set, valid_set, test_set, optimizer,
gpus=[0], batch_size=128)
solver.train(num_epoch=5)
solver.evaluate("valid")
任务 3:接触预测
我们要解决的第三个任务是预测折叠结构中任何一对残基是否接触,即进行接触预测。
我们首先通过构建数据集datasets.ProteinNet。该residue_position字段表示每个残基的 3D 坐标,该mask字段表示每个残基位置是否有效。两个字段都具有相同长度的蛋白质序列。
dataset = datasets.ProteinNet("~/protein-datasets/", atom_feature=None, bond_feature=None, residue_feature="default", transform=protein_view_transform)
train_set, valid_set, test_set = dataset.split()
print("Residue position: ", dataset[0]["graph"].residue_position[:3])
print("Valid mask: ", dataset[0]["graph"].mask[:3])
print("train samples: %d, valid samples: %d, test samples: %d" % (len(train_set), len(valid_set), len(test_set)))
数据结构
- “sequences”

- “targets”: 字段:‘valid_mask’和’tertiary’
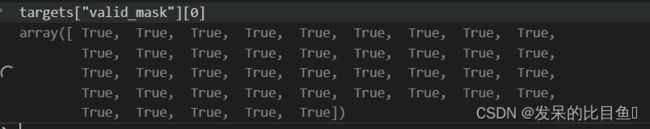
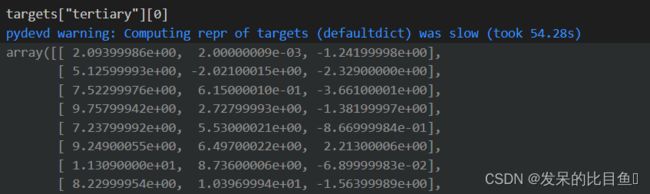
为了执行接触预测,我们将 CNN 编码器包装到tasks.ContactPrediction模块中,该模块在 CNN 上附加了一个任务特定的 MLP 预测头。如果欧几里得距离在 . 以内,则序列间隙大于 的两个残基gap被视为相互作用threshold。与以前的任务不同,max_length现在的任务中定义了最大截断长度,因为截断行为在接触预测任务的测试集上有所不同。对于测试集,为了节省内存,我们将测试序列按照 拆分成若干块max_length。
task = tasks.ContactPrediction(model, max_length=500, random_truncate=True, threshold=8.0, gap=6,
criterion="bce", metric=("accuracy", "prec@L5", "prec@5"), num_mlp_layer=2)
由于原始训练集包含大量样本,并且在此任务中只能使用小批量,因此我们使用原始训练集的一个子集和 1000 个样本进行训练。我们训练模型 1 个 epoch,大约需要 4 分钟,最后在验证集上对其进行评估。
from torch.utils import data as torch_data
optimizer = torch.optim.Adam(task.parameters(), lr=1e-4)
sub_train_set = torch_data.random_split(train_set, [1000, len(train_set) - 1000])[0]
solver = core.Engine(task, sub_train_set, valid_set, test_set, optimizer,
gpus=[0], batch_size=1)
solver.train(num_epoch=1)
solver.evaluate("valid")
任务 4:蛋白质-蛋白质相互作用 (PPI) 预测
我们考虑的第四个任务是预测两种相互作用蛋白质的结合亲和力,即执行PPI 亲和力预测。
我们首先通过 构建数据集datasets.PPIAffinity,其中每个样本都是一对蛋白质,并且与指示结合亲和力的连续标签相关联。由于我们现在需要对两种蛋白质进行转化,因此我们需要keys在转化函数中指定。
truncate_transform_ = transforms.TruncateProtein(max_length=200, keys=("graph1", "graph2"))
protein_view_transform_ = transforms.ProteinView(view="residue", keys=("graph1", "graph2"))
transform_ = transforms.Compose([truncate_transform_, protein_view_transform_])
dataset = datasets.PPIAffinity("~/protein-datasets/", atom_feature=None, bond_feature=None, residue_feature="default", transform=transform_)
train_set, valid_set, test_set = dataset.split()
print("The label of first sample: ", dataset[0][dataset.target_fields[0]])
print("train samples: %d, valid samples: %d, test samples: %d" % (len(train_set), len(valid_set), len(test_set)))
数据结构:
- “sequences”
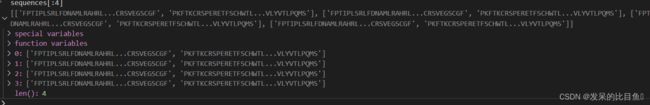
- “targets”, 字段 interaction

为了执行 PPI 亲和力预测,我们将 CNN 编码器包装到tasks.InteractionPrediction模块中,该模块将任务特定的 MLP 预测头附加到 CNN 上。
task = tasks.InteractionPrediction(model, task=dataset.tasks,
criterion="mse", metric=("mae", "rmse", "spearmanr"),
normalization=False, num_mlp_layer=2)
我们训练模型 10 个 epoch,大约需要 2 分钟,最后在验证集上对其进行评估。
optimizer = torch.optim.Adam(task.parameters(), lr=1e-4)
solver = core.Engine(task, train_set, valid_set, test_set, optimizer,
gpus=[0], batch_size=64)
solver.train(num_epoch=10)
solver.evaluate("valid")
任务 5:蛋白质-配体相互作用 (PLI) 预测
我们考虑的第四个任务是预测蛋白质和小分子(即配体)的结合亲和力。我们以BindingDB 上的 PLI 预测为例。
我们首先通过datasets.BindingDB构建数据集,其中每个样本都是一对蛋白质和配体,并且它与指示结合亲和力的连续标签相关联。我们使用该holdout_test集合进行测试。
truncate_transform_ = transforms.TruncateProtein(max_length=200, keys="graph1")
protein_view_transform_ = transforms.ProteinView(view="residue", keys="graph1")
transform_ = transforms.Compose([truncate_transform_, protein_view_transform_])
dataset = datasets.BindingDB("~/protein-datasets/", atom_feature=None, bond_feature=None, residue_feature="default", transform=transform_)
train_set, valid_set, test_set = dataset.split(["train", "valid", "holdout_test"])
print("The label of first sample: ", dataset[0][dataset.target_fields[0]])
print("train samples: %d, valid samples: %d, test samples: %d" % (len(train_set), len(valid_set), len(test_set)))
数据结构
- “sequences”

- “smiles”
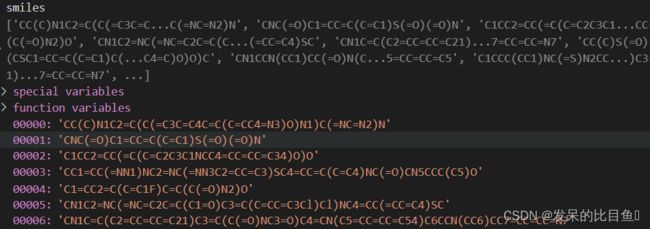
- “targets”: 字段affinity

为了执行 PLI 预测,我们需要一个额外的配体图编码器来提取配体的表示。我们定义了一个 4 层图同构网络 (GIN)作为配体图编码器。之后,我们将 CNN 编码器和 GIN 编码器包装到tasks.InteractionPrediction模块中,该模块将特定任务的 MLP 预测头附加到 CNN 和 GIN 上。
model2 = models.GIN(input_dim=66,
hidden_dims=[256, 256, 256, 256],
batch_norm=True, short_cut=True, concat_hidden=True)
task = tasks.InteractionPrediction(model, model2=model2, task=dataset.tasks,
criterion="mse", metric=("mae", "rmse", "spearmanr"),
normalization=False, num_mlp_layer=2)
我们训练模型 5 个 epoch,大约需要 3 分钟,最后在验证集上对其进行评估。
optimizer = torch.optim.Adam(task.parameters(), lr=1e-4)
solver = core.Engine(task, train_set, valid_set, test_set, optimizer,
gpus=[0], batch_size=16)
solver.train(num_epoch=5)
solver.evaluate("valid")