RNA 22. SCI 文章中基于表达估计恶性肿瘤组织的基质细胞和免疫细胞(ESTIMATE)

点击关注,桓峰基因
桓峰基因
生物信息分析,SCI文章撰写及生物信息基础知识学习:R语言学习,perl基础编程,linux系统命令,Python遇见更好的你
90篇原创内容
公众号
桓峰基因的教程不但教您怎么使用,还会定期分析一些相关的文章,学会教程只是基础,但是如果把分析结果整合到文章里面才是目的,觉得我们这些教程还不错,并且您安装我们的教程分析出来不错的结果发了文章记得告知我们,并在文章中感谢一下我们哦!
公司英文名称:Kyoho Gene Technology (Beijing) Co.,Ltd.
**如果您觉得这些确实没基础,需要专业的生信人员帮助分析,直接扫码加微信,我们24小时在线!!
**

每周线上直播课程,欢迎过来围观,扫码预约

前 言
恶性实体瘤组织不仅包括肿瘤细胞,还包括与肿瘤相关的正常上皮和基质细胞,免疫细胞和血管细胞。基质细胞被认为在肿瘤生长、疾病进展和耐药性中起重要作用。
浸润性免疫细胞的作用与环境有关,虽然浸润性T淋巴细胞的抗肿瘤作用在卵巢癌中已被观察到,但在结直肠癌中,肿瘤的生长、侵袭和转移与肿瘤的生长、侵袭和转移有关. 对肿瘤组织中与肿瘤相关的正常细胞的全面了解可能为肿瘤生物学的研究提供重要的见解,并有助于开发可靠的预后和预测模型。
ESTIMATE (Estimation of STromal and Immune cells in MAlignant Tumour tissues using Expression data): 利用癌症样本转录谱的独特性质来推断肿瘤细胞的内容以及不同的浸润正常细胞,该文章发表在 NC上,如下:

基本原理
浸润基质细胞和免疫细胞是肿瘤组织中正常细胞的主要组成部分,不仅在分子研究中干扰肿瘤信号,而且在肿瘤生物学中具有重要作用。在这里,我们描述了在恶性肿瘤中使用表达数据(ESTIMATE)估计基质细胞和免疫细胞的方法,该方法使用基因表达Signatures来推断肿瘤样本中基质细胞和免疫细胞的比例。评估分数与来自11种不同肿瘤类型样本的基于DNA拷贝数的肿瘤纯度相关,这些样本在安捷伦、Affymetrix平台上进行了分析,或基于RNA测序,可通过癌症基因组图谱获得。该预测的准确性进一步得到了在其他公共领域可用的3,809个转录谱的证实。ESTIMATE方法允许在基因组和转录组研究中考虑肿瘤相关正常细胞。其中算法流程表述的非常清晰,过来掉一些数据集,获得更优质的数据之后在进行差异分析筛选基因,如下:
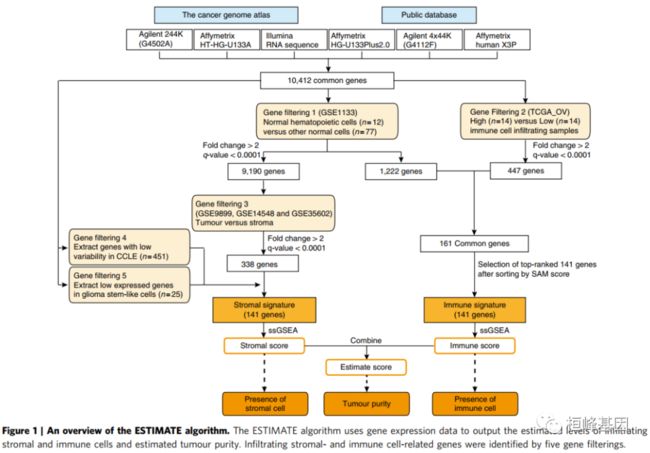
该软件重点研究基质细胞和免疫细胞,它们构成了肿瘤样本中主要的非肿瘤成分,并识别与肿瘤组织中基质细胞和免疫细胞浸润相关的特异性信号。通过进行单样本基因集富集分析(ssGSEA),通过计算基质和免疫评分来预测浸润基质和免疫细胞的水平,这些构成了在肿瘤组织中推断肿瘤纯度的 ESTIMATE score的基础。
实例解析
1. 软件安装
estimate 软件安装过程中需要我们进行网站的配置,该软件可以官网下载:
http://r-forge.r-project.org
if (!require(utils)) install.packages("utils")
library(utils)
if (!require(estimate)) install.packages("estimate", repos = "http://r-forge.r-project.org",
dependencies = TRUE)
library(estimate)
2. 数据读取
首先使用通过Affymetrix U133Plus2.0平台从10个卵巢癌样本中获得的数据。它具有由17,256个基因(行)和10个样本(列)组成的基因水平表达数据。其次,将每个微阵列平台的不同基因数量统一为10,412个共同基因。这些数据集包含的公共基因来自6个芯片平台:
1). Affymetrix HG-U133Plus2.0
2). Affymetrix HT-HG-U133A
3). Affymetrix Human X3P
4). Agilent 4x44K (G4112F)
5). Agilent G4502A
6). Illumina HiSeq RNA sequence
该算法使用HUGO GeneSymbols或Entrez基因ID。因此,输入数据中的行名称必须是基因SYMBOL或Entrez基因ID的矩阵,如下:
in.file <- system.file("extdata", "sample_input.txt", package = "estimate") #输入文件
read.table(in.file)[1:4, 1:4]
## s516 s518 s519 s520
## C9orf152 4.881540 4.575656 3.739469 3.695996
## ELMO2 7.298054 7.555440 7.533202 7.382355
## CREB3L1 5.569164 5.700406 5.959730 5.770007
## RPS11 13.389937 13.848820 13.642862 13.654622
dim(read.table(in.file))
## [1] 17256 10
outfile2E <- "ESTIMATE_input.gct"#生成ESTIMATE 的输入文件
outputGCT(in.file, outfile2E)
3. 免疫细胞估计
计算基质细胞和免疫细胞评分,分别代表肿瘤组织中基质和免疫细胞的存在。该功能将每个平台的不同数量的基因与10,412个普通基因相结合,如下:
filterCommonGenes(input.f = in.file, output.f = outfile2E, id = "GeneSymbol")
## [1] "Merged dataset includes 10412 genes (0 mismatched)."
这个功能计算基质,免疫,并估计得分每个样本使用基因表达数据,如下:
estimateScore("ESTIMATE_input.gct", "ESTIMATE_score.gct")
## [1] "1 gene set: StromalSignature overlap= 141"
## [1] "2 gene set: ImmuneSignature overlap= 141"
根据ESTIMATE score绘制肿瘤纯度,结果如下:
plotPurity(scores = "ESTIMATE_score.gct", samples = "s516")

保存评分结果,如下:
# 将评分保存为txt格式
scores = read.table("ESTIMATE_score.gct", skip = 2, header = T)
rownames(scores) = scores[, 1]
scores = t(scores[, 3:ncol(scores)])
scores
## StromalScore ImmuneScore ESTIMATEScore TumorPurity
## s516 -281.81487 171.5411 -110.2737 0.8316075
## s518 -426.14692 105.3890 -320.7580 0.8483668
## s519 -57.14977 -365.2374 -422.3871 0.8561698
## s520 1938.82379 2339.0707 4277.8944 0.3314725
## s521 -671.64710 147.6183 -524.0288 0.8637832
## s522 1458.13837 1176.8159 2634.9543 0.5472110
## s523 -268.89216 -928.4953 -1197.3875 0.9092887
## s525 973.42289 1320.0869 2293.5098 0.5884565
## s526 552.64161 2162.4612 2715.1029 0.5373262
## s527 -709.33568 1312.8416 603.5059 0.7689656
write.table(scores, file = "ESTIMATE_score.txt", quote = F, sep = "\t")
结果解读
我们得到结果之后,怎么才能应用到文章中,这里我们选择公众号有一期分享过的文章:IF:7+ 免疫微环境中的免疫基因标记和免疫类型与胶质瘤的预后相关,这里根据基于ssGSEA scores进行无监督聚类之后,在从免疫细胞方向看分组的准确性,但是其实ssGSEA 的打分与 ESTIMATE 的估计值是高度一致的,因此其结果也不奇怪,并没有说明任何问题。那么我们看下是怎么应用这种办法的,首先在Methods里面写道:
The ESTIMATE algorithm [doi:10.1038/ncomms3612] was used to analyze the Immune Score, Stromal Score, ESTIMATE Score, and tumor purity.
再看结果里面是这样提到该分析部分,如下:
Using the CGGA cohort, first, we performed an unsupervised clustering analysis and identified three distinct immunesubgroup patterns based on the ssGSEA scores of 25 immunerelated gene sets (Figures 1A, S1A–E). The three immunesubgroups were correspondingly termed as the immune-H (high) (n = 226), immune-M (media) (n = 185), and immuneL (low) (n=338). The immune-H subgroup had the highest values of ESTIMATE Score, Immune Score, and Stromal Score; it was also associated with the lowest tumor purity (Figures 1B–E). Notably, these results indicated that the immune-H subgroup consisted of the highest number of immune cells and stromal cells, while the immune-L subgroup consisted of the highest number of tumor cells; these values in the immune-M subgroup were between in those for the immune-H and immune-L subgroups.
我们再看下分析结果展示的图表 Figures 1B–E,这里我把图表的图注也显示出来,这样大家就知道怎么才能更好的使用这个分析模块,如下:

References
-
Subramanian A, et al.Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles.Proc Natl Acad Sci U S A 2005, 102:15545-15550.
-
Barbie DA, et al.Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1.Nature 2009, 462:108-112.
-
Verhaak RG, et al.Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1.Cancer Cell 2010, 17:98-110.
-
Carter SL, et al.Absolute quantification of somatic DNA alterations in human cancer.Nat Biotechnol 2012, 30:413-421.
-
Wang XX, Cao H, Zhai Y, et al. Immune Gene Signatures and Immunotypes in Immune Microenvironment Are Associated With Glioma Prognose. Front Immunol. 2022;13:823910. Published 2022 Apr 14. doi:10.3389/fimmu.2022.823910