生信学习——基于R的统计习题(附详细答案解读)
题目目录
- 基础概念
-
- 1. 载入R中自带的数据集 iris,指出其每列是定性还是定量数据
- 2. 对数据集 iris的所有定量数据列计算集中趋势指标:众数、分位数和平均数
- 3. 对数据集 iris的所有定性数据列计算水平及频次
- 4. 对数据集 iris的所有定量数据列计算离散趋势指标:方差和标准差等
- 5. 计算数据集 iris的前两列变量的相关性,提示cor函数可以选择3种methods
- 6. 对数据集 iris的所有定量数据列内部z-score标准化,并计算标准化后每列的平均值和标准差
- 7. 计算列内部zcore标准化后 iris的前两列变量的相关性
- 8. 根据数据集 iris的第五列拆分数据集后重复上面的Q2到Q7问题
- 9. 载入R中自带的数据集 mtcars,重复上面的Q1到Q7个问题
- 10. 载入r包airway并且通过assay函数拿到其表达矩阵后计算每列之间的相关性
- 表达矩阵相关
-
- 1. 把RNAseq_expr第一列全部加1后取log2后计算平均值和标准差
- 2. 根据上一步得到平均值和标准差生成同样个数的随机的正态分布数值
- 3. 删除RNAseq_expr第一列低于5的数据后,重复Q1和Q2
- 4. 基于Q3对RNAseq_expr的第一列和第二列进行T检验
- 5. 取RNAseq_expr行之和最大的那一行根据分组矩阵进行T检验
- 6. 取RNAseq_expr的MAD(绝对中位差)最大的那一行根据分组矩阵进行T检验
- 7. 对RNAseq_expr全部加1后取log2后重复Q5和Q6
- 8. 取RNAseq_expr矩阵的MAD最高的100行,对列和行分别进行层次聚类
- 9. 取RNAseq_expr矩阵的SD最高的100行,对列和行分别进行层次聚类
- 10. 对Q8矩阵按照行和列分别归一化并且热图可视化
- 统计检验相关
-
- 1. 对e1每一行独立根据分组矩阵进行T检验,检查为什么有些行T检验失败
- 2. 找出T检验失败的行并且从e1矩阵剔除掉
- 3. 对过滤后的e1矩阵进行每一行独立根据分组矩阵进行T检验
- 4. 对e1矩阵进行加1后log2的归一化命名为e2再对每一行独立根据分组矩阵进行T检验
- 5. 对e1,e2的T检验P值做相关性分析
写在前面——本文是介绍了一些统计学的基础知识,以及如何用R语言实现。旨在熟练使用编程语言解决实际问题。统计学是一门复杂的学科,还需更加系统的学习。下方 生统基础是大佬写的StartQuest的读书笔记,能够帮助快速理解生物统计学。
题目原文:http://www.bio-info-trainee.com/4385.html
参考答案:https://www.jianshu.com/p/1c0cbf1bfb75
参考答案:https://hc1023.github.io/2019/09/27/R-statistic/
生统基础:http://rvdsd.top/categories/%E7%94%9F%E7%BB%9F%E4%B9%8BStatQuest/
基础概念
1. 载入R中自带的数据集 iris,指出其每列是定性还是定量数据
colnames(iris)
head(iris)

基础概念讲解:https://mp.weixin.qq.com/s/OtB2h6f00U2SRZLzveJKfQ
前四个为定量变量,最后一个为定性变量
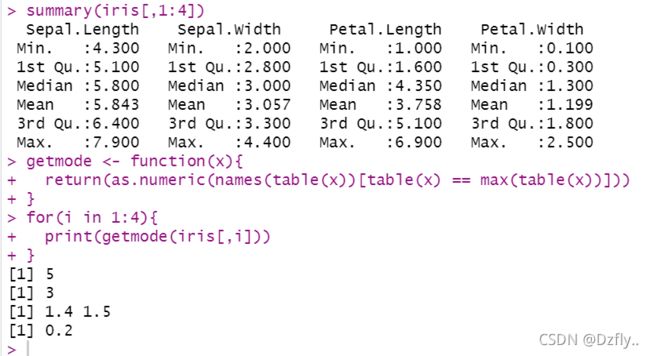
2. 对数据集 iris的所有定量数据列计算集中趋势指标:众数、分位数和平均数
# 求前四个变量的最大值、最小值、分位数和平均数
summary(iris[,1:4])
# R中无直接求众数的函数
# 法一
# tmp <- table(iris[,1])
# index <- which.max(tmp)
# tmp[index]
# 法二
getmode <- function(x){
return(as.numeric(names(table(x))[table(x) == max(table(x))]))
}
# table(iris[,1]) == max(table(iris[,1]))
# names(table(iris[,1]))
# names(table(iris[,1]))[table(iris[,1]) == max(table(iris[,1]))]
for(i in 1:4){
print(getmode(iris[,i]))
}
# 第三列有两个众数
3. 对数据集 iris的所有定性数据列计算水平及频次
summary(iris[,5])
#output#
setosa versicolor virginica
50 50 50
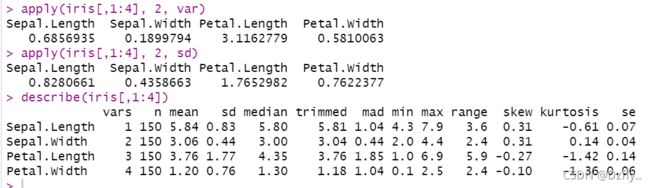
4. 对数据集 iris的所有定量数据列计算离散趋势指标:方差和标准差等
# 方差
apply(iris[,1:4], 2, var)
# 标准差
apply(iris[,1:4], 2, sd)
# 通过psych包中的describe()计算描述性统计量
# 输出结果依次为:非缺失值的数量、平均数、标准差、中位数、截尾均值、绝对中位差、最小值、最大值、值域、偏度、峰度和平均值的标准误
# install.packages("psych")
library(psych)
describe(iris[,1:4])
5. 计算数据集 iris的前两列变量的相关性,提示cor函数可以选择3种methods
cor(iris[,1:2], method = "pearson")
cor(iris[,1:2], method = "spearman")
cor(iris[,1:2], method = "kendall")
Pearson积差相关系数衡量了两个定量变量之间的线性相关程度(默认值)
Spearman等级相关系数则衡量分级定序变量之间的相关程度
Kendall’sTau相关系数也是一种非参数的等级相关度量

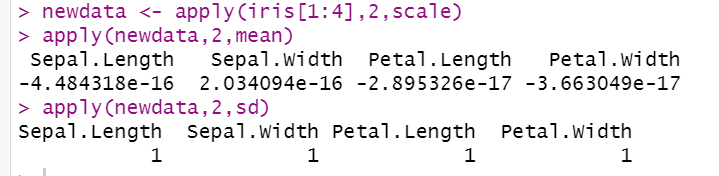
6. 对数据集 iris的所有定量数据列内部z-score标准化,并计算标准化后每列的平均值和标准差
newdata <- apply(iris[1:4],2,scale)
apply(newdata,2,mean)
apply(newdata,2,sd)

图源:https://blog.csdn.net/Orange_Spotty_Cat/article/details/80312154
scale(x, center = TRUE, scale = TRUE)
center和scale默认为真,center为真表示数据中心化,scale为真表示数据标准化
数据中心化是指:变量减去它的均值
数据标准化是指:数值减去均值,再除以标准差
所以不难理解,对数据的每列调用scale就完成了z-score标准化
标准化之后的数据均值为0,标准差为1(上面的均值结果近似于0,也算为0吗?)
参考:https://zhuanlan.zhihu.com/p/32482328
7. 计算列内部zcore标准化后 iris的前两列变量的相关性
cor(newdata[,1:2], method = "pearson")
cor(newdata[,1:2], method = "spearman")
cor(newdata[,1:2], method = "kendall")

与第5题结果相同,标准化后的数据相关性不变
8. 根据数据集 iris的第五列拆分数据集后重复上面的Q2到Q7问题
# 划分数据集
table(iris$Species)
setosa <- iris[iris$Species=="setosa",]
head(setosa)
versicolor <- iris[iris$Species=="versicolor",]
virginica <- iris[iris$Species=="virginica",]
# 写个无脑输出函数
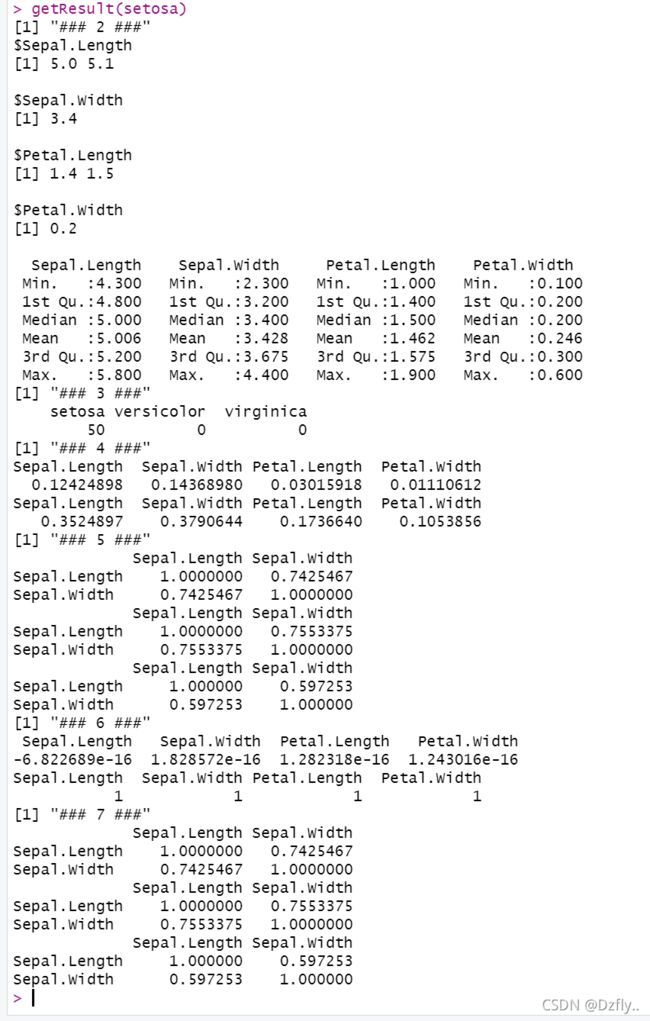
getResult <- function(x){
print("### 2 ###")
print(apply(x[,1:4], 2, getmode))
print(summary(x[,1:4]))
print("### 3 ###")
print(summary(x[,5]))
print("### 4 ###")
print(apply(x[,1:4], 2, var))
print(apply(x[,1:4], 2, sd))
describe(x[,1:4])
print("### 5 ###")
print(cor(x[,1:2], method = "pearson"))
print(cor(x[,1:2], method = "spearman"))
print(cor(x[,1:2], method = "kendall"))
print("### 6 ###")
newx <- apply(x[1:4],2,scale)
print(apply(newx,2,mean))
print(apply(newx,2,sd))
print("### 7 ###")
print(cor(newx[,1:2], method = "pearson"))
print(cor(newx[,1:2], method = "spearman"))
print(cor(newx[,1:2], method = "kendall"))
}
getResult(setosa)
getResult(versicolor)
getResult(virginica)
9. 载入R中自带的数据集 mtcars,重复上面的Q1到Q7个问题
# q1
> colnames(mtcars)
[1] "mpg" "cyl" "disp" "hp" "drat" "wt" "qsec" "vs" "am" "gear" "carb"
# mtcars每列数据的含义
Format
A data frame with 32 observations on 11 (numeric) variables.
[, 1] mpg Miles/(US) gallon
[, 2] cyl Number of cylinders
[, 3] disp Displacement (cu.in.)
[, 4] hp Gross horsepower
[, 5] drat Rear axle ratio
[, 6] wt Weight (1000 lbs)
[, 7] qsec 1/4 mile time
[, 8] vs Engine (0 = V-shaped, 1 = straight)
[, 9] am Transmission (0 = automatic, 1 = manual)
[,10] gear Number of forward gears
[,11] carb Number of carburetors
# 定量变量一般使用具体的数字表示,定性变量也称分类变量,可以用来分类
# 在这里,8和9明显为定性变量
# 其余列为定量变量
# q2-q7
# 将定量变量取出,放在一起
newmtcars <- cbind(mtcars[,1:7],mtcars[10:11])
# 没设置变量,直接暴力输出
getMtcars <- function(){
print("### 2 ###")
print(apply(newmtcars, 2, getmode))
print(summary(newmtcars))
print("### 3 ###")
print(apply(mtcars[8:9], 2, table))
print("### 4 ###")
print(apply(newmtcars, 2, var))
print(apply(newmtcars, 2, sd))
describe(newmtcars)
print("### 5 ###")
print(cor(mtcars[,1:2], method = "pearson"))
print(cor(mtcars[,1:2], method = "spearman"))
print(cor(mtcars[,1:2], method = "kendall"))
print("### 6 ###")
newx <- apply(newmtcars,2,scale)
print(apply(newx,2,mean))
print(apply(newx,2,sd))
print("### 7 ###")
print(cor(newx[,1:2], method = "pearson"))
print(cor(newx[,1:2], method = "spearman"))
print(cor(newx[,1:2], method = "kendall"))
}
getMtcars()
# mpg和cyl的负相关性还是很强的

10. 载入r包airway并且通过assay函数拿到其表达矩阵后计算每列之间的相关性
summarizedexperiment详细说明:https://bioconductor.org/packages/release/bioc/vignettes/SummarizedExperiment/inst/doc/SummarizedExperiment.html#anatomy-of-a-summarizedexperiment
其中关于airway的描述:
该软件包包含来自气道平滑肌每个基因的读取计数的 RNA-Seq实验的示例数据集。这些数据存储在一个对象中,该对象包含 8 个不同的实验和分析 64,102 个基因转录本。
人性化解读:https://www.dazhuanlan.com/nby/topics/1127479
rm(list=ls())
options(stringsAsFactors = F)
# BiocManager::install("airway")
library(airway)
data("airway")
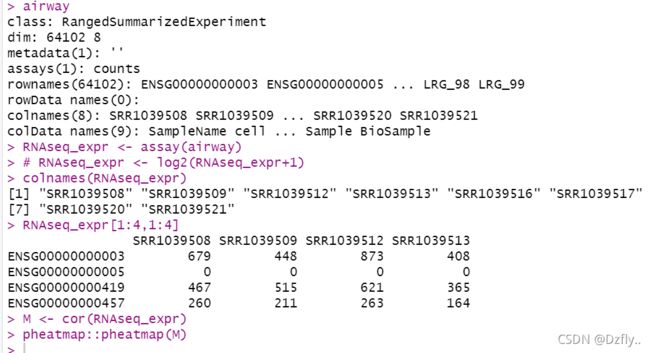
airway
# 获取实验数据
RNAseq_expr <- assay(airway)
colnames(RNAseq_expr)
RNAseq_expr[1:4,1:4]
# RNAseq_gl <- colData(airway)[,3]
# table(RNAseq_gl)
# 计算相关性
M <- cor(RNAseq_expr)
pheatmap::pheatmap(M)

表达矩阵相关
1. 把RNAseq_expr第一列全部加1后取log2后计算平均值和标准差
# 清空环境,重新取一下数据
rm(list=ls())
options(stringsAsFactors = F)
library(airway)
data("airway")
RNAseq_expr <- assay(airway)
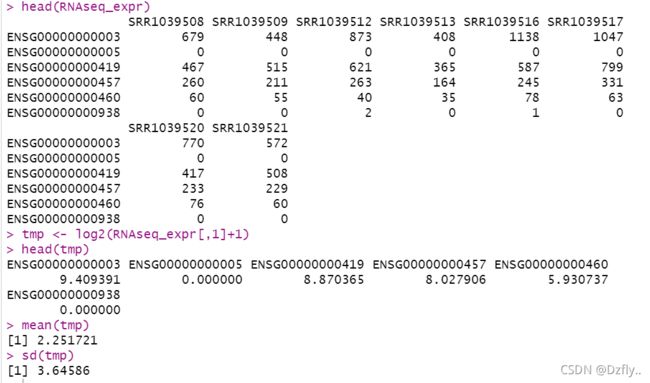
head(RNAseq_expr)
# +1是因为0没法取对数
tmp <- log2(RNAseq_expr[,1]+1)
head(tmp)
mean(tmp)
sd(tmp)
2. 根据上一步得到平均值和标准差生成同样个数的随机的正态分布数值
# dnorm给出密度,pnorm给出分布函数,qnorm给出分位数函数,rnorm产生随机偏差
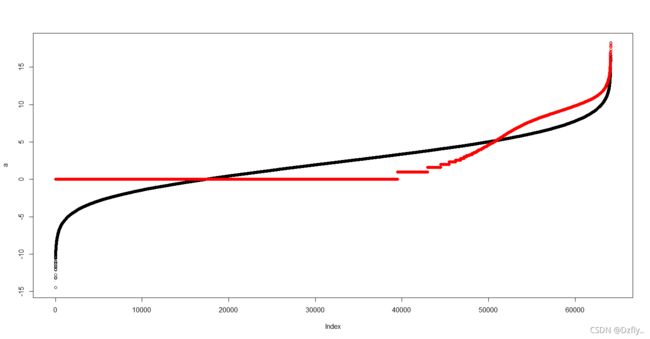
a <- rnorm(length(tmp),mean = mean(tmp),sd = sd(tmp))
# 升序排序
a <- sort(a)
plot(a)
# 跟处理前的数据分布对比一下
points(sort(tmp), col = "red")
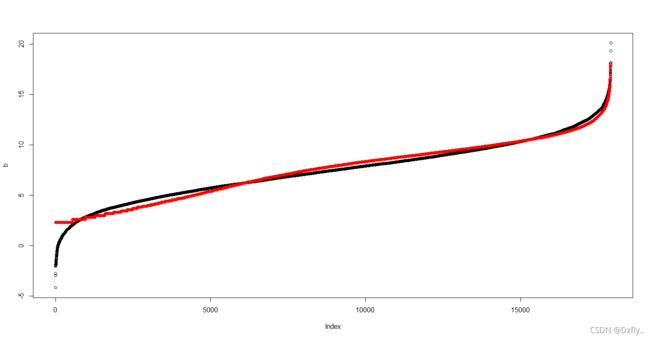
3. 删除RNAseq_expr第一列低于5的数据后,重复Q1和Q2
# 删除对应数据
NewRNAseq_expr <- RNAseq_expr[RNAseq_expr[,1]>=5, ]
# Q1
# 没有+1是因为数据不存在0了
tmp <- log2(NewRNAseq_expr[,1])
head(tmp)
mean(tmp)
sd(tmp)
# Q2
b <- rnorm(length(tmp), mean = mean(tmp), sd = sd(tmp))
b <- sort(b)
plot(b)
points(sort(tmp), col = "red")
# 可以看到,筛选过的数据就很接近正态分布了
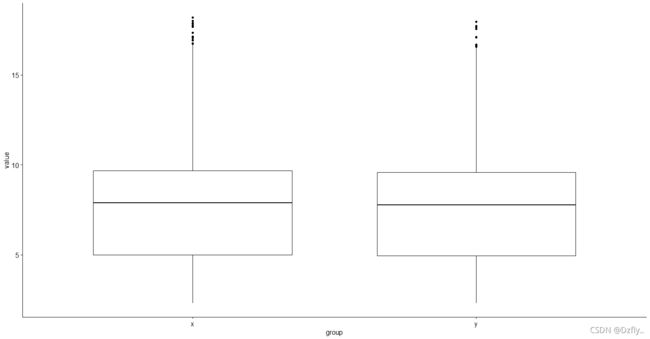
4. 基于Q3对RNAseq_expr的第一列和第二列进行T检验
# 选择数据进行T检验
x <- RNAseq_expr[RNAseq_expr[,1]>=5, ]
y <- RNAseq_expr[RNAseq_expr[,2]>=5, ]
x <- log2(x[,1])
y <- log2(y[,2])
t.test(x,y)
# 绘制箱图,查看数据分布情况
library(ggplot2)
library(ggpubr)
df <- data.frame(value=c(x,y),group=c(rep('x',length(x)),rep('y',length(y))))
ggboxplot(df, y = "value", x = "group")

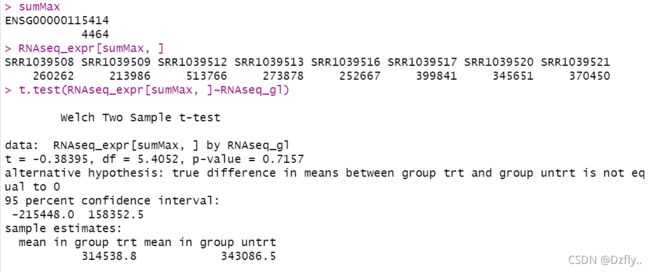
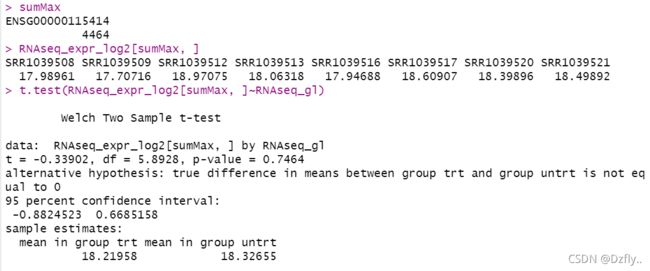
5. 取RNAseq_expr行之和最大的那一行根据分组矩阵进行T检验
# 找到和最大的那一行
sumMax <- which.max(rowSums(RNAseq_expr))
sumMax
RNAseq_expr[sumMax, ]
# 分组情况
RNAseq_gl <- colData(airway)[,3]
table(RNAseq_gl)
# “~”左边的是一个数值型变量,右边是一个二分变量
t.test(RNAseq_expr[sumMax, ]~RNAseq_gl)
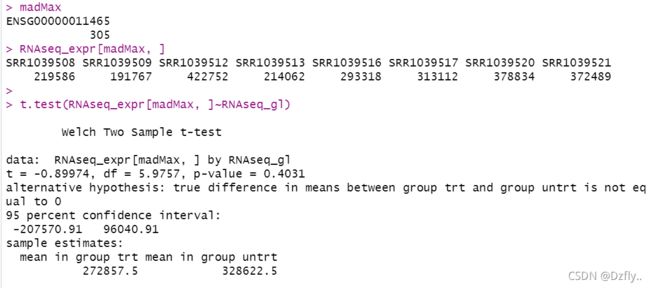
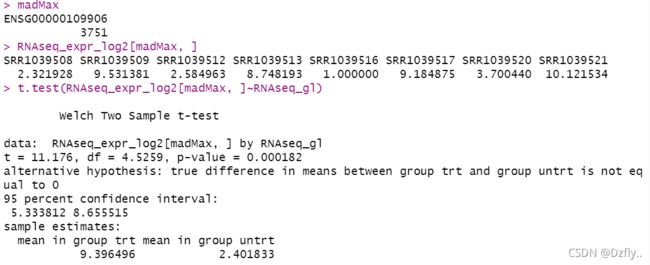
6. 取RNAseq_expr的MAD(绝对中位差)最大的那一行根据分组矩阵进行T检验
绝对中位差实际求法是用原数据减去中位数后得到的新数据的绝对值的中位数。但绝对中位差常用来估计标准差,估计标准差=1.4826*绝对中位差。R语言中返回的是估计的标准差。
madMax <- which.max(apply(RNAseq_expr, 1, mad))
madMax
RNAseq_expr[madMax, ]
t.test(RNAseq_expr[madMax, ]~RNAseq_gl)
7. 对RNAseq_expr全部加1后取log2后重复Q5和Q6
RNAseq_expr_log2 <- log2(RNAseq_expr+1)
# Q5
# 基因没变,P值变化不大
sumMax <- which.max(rowSums(RNAseq_expr_log2))
sumMax
RNAseq_expr_log2[sumMax, ]
t.test(RNAseq_expr_log2[sumMax, ]~RNAseq_gl)
# Q6
# 基因变了,P值变化很大
# mad可以反映数据的波动情况,只是粗略的一个估计值,辅助判断
madMax <- which.max(apply(RNAseq_expr_log2, 1, mad))
madMax
RNAseq_expr_log2[madMax, ]
t.test(RNAseq_expr_log2[madMax, ]~RNAseq_gl)
8. 取RNAseq_expr矩阵的MAD最高的100行,对列和行分别进行层次聚类
# 取log2之后的数据
cg1 <- names(tail(sort(apply(RNAseq_expr_log2, 1, mad)), 100))
dat1 <- RNAseq_expr_log2[cg1, ]
# 原始数据
cg2 <- names(tail(sort(apply(RNAseq_expr, 1, mad)), 100))
dat2 <- RNAseq_expr[cg2, ]
# 对行进行聚类
plot(hclust(dist(t(dat1))))
plot(hclust(dist(t(dat2))))
# 对列进行聚类
plot(hclust(dist(dat1)))
plot(hclust(dist(dat2)))
# 聚类之后的图不知道怎么解读...挖坑待填...

9. 取RNAseq_expr矩阵的SD最高的100行,对列和行分别进行层次聚类
cg <- names(tail(sort(apply(RNAseq_expr_log2, 1, sd)), 100))
dat <- RNAseq_expr_log2[cg, ]
plot(hclust(dist(t(dat))))
plot(hclust(dist(dat)))
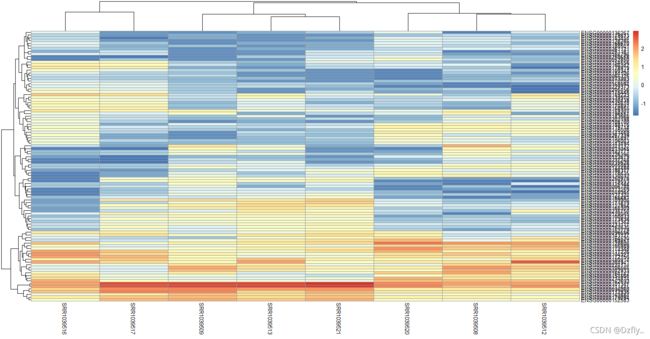
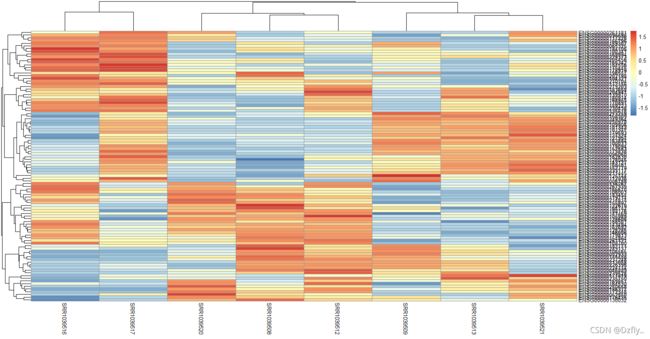
10. 对Q8矩阵按照行和列分别归一化并且热图可视化
# 紧接Q8
pheatmap::pheatmap(scale(dat1))
pheatmap::pheatmap(t(scale(t(dat1))))
# 图很乱,想看清楚还需再处理一下
统计检验相关
这里需要对前面的RNAseq_expr矩阵进行一定程度的过滤
rm(list=ls())
options(stringsAsFactors = F)
library(airway)
data("airway")
RNAseq_expr <- assay(airway)
colnames(RNAseq_expr)
dim(RNAseq_expr)
RNAseq_gl <- colData(airway)[,3]
table(RNAseq_gl)
# 删除 每列相加,和小于等于1 的数据
e1 <- RNAseq_expr[apply(RNAseq_expr,1,function(x) sum(x>0)>1),]
dim(e1)
#output#
> dim(RNAseq_expr)
[1] 64102 8
> dim(e1)
[1] 28877 8
上面的 e1 矩阵下面的习题就用得到。
1. 对e1每一行独立根据分组矩阵进行T检验,检查为什么有些行T检验失败
apply(e1, 1, function(x){
t.test(x ~ RNAseq_gl)$p.value
})
#output#
Error in t.test.default(x = c(SRR1039509 = 1L, SRR1039513 = 1L, SRR1039517 = 1L, :
data are essentially constant
# 使用二分法手动查找出错的位置
> t.test(e1[4257,] ~ RNAseq_gl)$p.value
Error in t.test.default(x = c(SRR1039509 = 1L, SRR1039513 = 1L, SRR1039517 = 1L, :
data are essentially constant
> rownames(e1)[4257]
[1] "ENSG00000116852"
> e1[4257,]
SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517 SRR1039520 SRR1039521
0 1 0 1 0 1 0 1
# 通过观察数据可以发现,报错应该是因为相同组的数据都一样,没法进行T检验
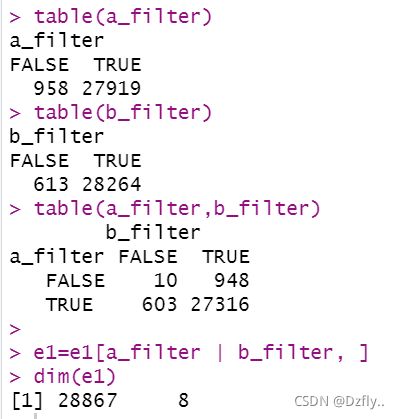
2. 找出T检验失败的行并且从e1矩阵剔除掉
# 分类
e1_a <- e1[,RNAseq_gl=='trt']
e1_b <- e1[,RNAseq_gl=='untrt']
# 筛选,如果数据相同的话,行标准差为0
a_filter <- apply(e1_a, 1, function(x) sd(x)>0)
b_filter <- apply(e1_b, 1, function(x) sd(x)>0)
# 看一下筛选的结果
table(a_filter)
table(b_filter)
table(a_filter,b_filter)
# 去除同时为FALSE的值
e1 <- e1[a_filter | b_filter, ]
3. 对过滤后的e1矩阵进行每一行独立根据分组矩阵进行T检验
p1 <- apply(e1, 1, function(x){
t.test(x~RNAseq_gl)$p.value
})
head(p1)
#output#
ENSG00000000003 ENSG00000000419 ENSG00000000457 ENSG00000000460 ENSG00000000938
0.2208266 0.8275543 0.6747468 0.3841246 0.2151699
ENSG00000000971
0.4949860
4. 对e1矩阵进行加1后log2的归一化命名为e2再对每一行独立根据分组矩阵进行T检验
e2 <- log2(e1+1)
p2 <- apply(e2, 1, function(x){
t.test(x ~ RNAseq_gl)$p.value
})
head(p2)
#output#
ENSG00000000003 ENSG00000000419 ENSG00000000457 ENSG00000000460 ENSG00000000938
0.1705491 0.9265533 0.5472763 0.4442560 0.1975805
ENSG00000000971
0.6024837
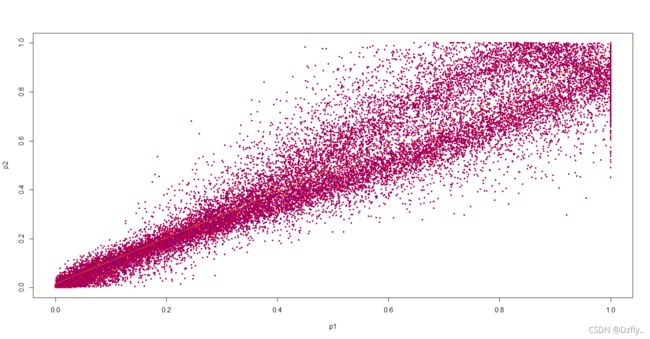
5. 对e1,e2的T检验P值做相关性分析
plot(p1, p2, cex = 0.5, pch = 21, col = "red", bg = "blue")
# 画出最小二乘拟合直线
abline(lsfit(p1, p2), col = "yellow", lty = 3)
cor(p1,p2)
#output#
[1] 0.9470391