- Matlab S-Function模块简谈
Captain cool290
matlab
1.单个输出形式的m脚本文件如何编写functiony=fcn()y=u;最简单的例子:就是输出等于输入点一下标题栏:gotodiagram就可以回到模型界面;EditData可以修改数据类型2.多个输出形式的m脚本如何编写function[y1,y2]=fcn(u1,u2)y1=u1+u2;y2=u2-u1;这样就可以多个输入和输出了。mamatlab3.注意点:S-Function中的变量u是
- Ai时代初期,人类文明的多纬度演进方向分析
Ai度
人工智能
在AI时代初期,文明的演进呈现出多维度、跨领域的突破性特征,结合最新研究进展与实践案例,其深层变革可进一步细化为以下六大维度:一、技术平权与生产要素重构AI技术通过算力跃迁与认知革命重构生产要素。例如,华为昇腾芯片使县域政务系统获得省级决策能力,特斯拉工厂的机械臂实现0.8秒完成车身焊接,而量子-经典混合算法将药物分子模拟效率提升1200倍。这种技术平权运动正推动全球劳动生产率提升30%,同时催生
- pear-admin-boot开发框架使用记录(三)
后青春期的诗go
经验分享javaspringbootspringlog4jmybatis
一、实现部门选择操作用于从组织架构里选择出部门的操作,如开发日志管理模块,创建人新增日志时可以通过选择框选择相应共享的部门。数据库表调整在数据表添加2个字段:sharedeptid共享部门idvarcharsharedeptname共享部门名称varchar前端html页面调整页面添加如下代码:共享部门前端JS调整添加如下代码:letdtree=layui.dtree;dtree.renderSe
- 基于koajsAdmin+mongodb的后台管理快速开发框架安装运行记录
后青春期的诗go
经验分享mongodb数据库node.jsvue.jselementui
前置操作下载源码源码地址:https://gitee.com/zhoushuigui/koajs-admin安装mongodb数据库并连接安装yarnnpminstallyarn-g安装nodemonnpminstallnodemon-g前端运行安装依赖进入项目根目录,在命令行执行如下命令安装依赖:yarn--registry=https://registry.npmmirror.com启动服务y
- 源代码防泄密和安全上外网的关联
cnsinda_sdc
源码安全源代码加密信息安全网络安全服务器源代码防泄露
在数字化办公的时代,企业员工需要频繁访问互联网以获取信息、进行沟通和协作。然而,互联网的开放性也带来了诸多安全风险,如恶意软件、网络攻击、数据泄露等。SPN沙盒作为一种先进的安全上网解决方案,为企业提供了一种安全、可控的上网方式。一、系统构成与部署(一)管理端管理端是SPN沙盒系统的控制中心,负责对整个沙盒系统进行管理控制。它具备以下功能:系统配置与策略管理:管理员可以通过管理端对SPN沙盒系统进
- Redis 详解
z小天才b
Redisredis数据库缓存
1.NoSQL的核心概念和应用场景核心概念NoSQL(NotOnlySQL):一类非关系型数据库的统称,专为处理大规模数据存储而设计特点:高扩展性、高性能、灵活的数据模型、分布式架构CAP理论:一致性(Consistency)、可用性(Availability)、分区容错性(Partitiontolerance),NoSQL通常优先保证AP或CP主要类型键值存储:Redis,Memcached文档
- 【BERT和GPT的区别】
调皮的芋头
人工智能深度学习机器学习bertgpt
BERT采用完形填空(MaskedLanguageModeling,MLM)与GPT采用自回归生成(AutoregressiveGeneration)的差异,本质源于两者对语言建模的不同哲学导向与技术目标的根本分歧。这种选择不仅塑造了模型的架构特性,更决定了其应用边界与能力上限。以下从语言建模本质、任务适配性、技术约束及后续影响四个维度深入剖析:一、语言建模的本质差异1.BERT的“全知视角”与全
- Laravel 8 项目基于 PHP 8 与 Nginx 的线上部署全攻略
你华还是你华
laravel上线级项目phplaravelnginx
本文目录前言一、服务器1.1购买与选型1.2服务器配置安装php8二、项目上线2.1git关联2.2安装项目依赖2.3项目配置2.3.1基础配置2.3.2数据库及表配置与创建2.3.3Navicat连接Mysql2.3.4运行seeder进行数据填充2.3.5Nginx配置与报错处理三、项目成功调用API示例四、自动配置https证书4.1Certbot概述4.2配置证书4.3自动更新证书4.4效
- 微信小程序云开发实现登录功能
Bilkan-studio
微信小程序小程序前端
使用云开发数据库实现登录功能,多的不说了直接看代码登录功能代码段WXML代码账号密码登录WXSS代码page{width:100%;height:100%;direction:ltr;}.waikuang{width:100%;height:100%;display:flex;align-items:center;justify-content:center;flex-direction:colu
- python 读取内存_python内存读写
weixin_39981360
python读取内存
广告关闭腾讯云11.11云上盛惠,精选热门产品助力上云,云服务器首年88元起,买的越多返的越多,最高返5000元!也就是说,所有的解释器可以同时读写数据,在一个解释器中对数据做出的修改会自动反映到其他解释器上。虽然还需要一些额外的步骤来处理同步问题,但是有时候可以使用这种方法作为通过管道或者socket传输数据的替代方案。以上这篇python内存映射文件读写方式就是小编分享给大家的全部内容了,希望
- 基于Python的智能决策支持系统:实现智能化决策的关键要素
AI天才研究院
DeepSeekR1&大数据AI人工智能大模型自然语言处理人工智能语言模型编程实践开发语言架构设计
文章目录基于Python的智能决策支持系统:实现智能化决策的关键要素11.背景介绍2.核心概念与联系数据收集与预处理模型构建与训练决策规则生成与优化决策结果评估与反馈3.核心算法原理具体操作步骤数据挖掘算法机器学习算法优化算法4.数学模型和公式详细讲解举例说明线性回归模型最小二乘法5.项目实践:代码实例和详细解释说明6.实际应用场景金融领域医疗领域供应链管理智能制造7.工具和资源推荐编程语言和开发
- 如何返回工具执行的工件
shuoac
windowsmicrosoftpython
工具是在AI模型中被调用并将输出反馈给模型的实用程序。有时,我们希望将工具执行的工件,例如自定义对象、数据帧或图像,传递给链或代理中的下游组件,但不希望向模型本身暴露这些工件。为了实现这一点,Tool和ToolMessage接口提供了一个机制,以区分提供给模型的信息(ToolMessage.content)和供外部使用的信息(ToolMessage.artifact)。技术背景介绍在AI开发中,工
- hive 使用oracle数据库
sardtass
hadoophive开源项目
hive使用oracle作为数据源,导入数据使用sqoop或kettle或自己写代码(淘宝的开源项目中有一个xdata就是淘宝自己写的)。感觉sqoop比kettle快多了,淘宝的xdata没用过。hive默认使用derby作为存储表信息的数据库,默认在哪启动就在哪建一个metadata_db文件放数据,可以在conf下的hive-site.xml中配置为一个固定的位置,这样不论在哪启动都可以了。
- 常州 d8
Rikka's_qwq
算法c++学习
好难啊哈哈哈大家考得好像都不是很好中午刚出成绩就发了动态了我也真是被自己无语到了t1测试样例时输出的数据没注释掉爆零t2freopen注释掉了爆零啊哈哈t1虽然我写的是最朴素的做法...但好歹能骗40分呢给我炸了正解是这样的#include#defineintlonglong#definedoublelongdouble#definephi(sqrt(5)+1)/2usingnamespacest
- leetcode刷题(javaScript)——栈、单调栈相关场景题总结
三月的一天
Leetcode刷题技巧总结javascriptleetcodelinux
在LeetCode刷题中,栈是一个常用的数据结构,可以帮助解决很多问题。以下是一些需要使用栈的方法,以及单调栈的应用场景:栈的使用技巧:栈常用于解决与括号匹配相关的问题,如括号序列的有效性、最长有效括号等。栈也常用于解决逆波兰表达式、表达式求值等与计算相关的问题。栈可以用于解决深度优先搜索(DFS)中的回溯问题,如组合、排列等。栈还可以用于解决某些需要“后进先出”(LIFO)特性的问题,如某些遍历
- Starrocks使用中一些总结
WYRM_GOLD
Starrocks数据库数据仓库数据库开发
1、实时写入的注意事项数据表如果是每天写入,要创建分区(推荐按天分区)。表模型选择更新模型查询的维度列,整数类型列放在前面,有利于快速的查询表分区要设置过期时间,尽可能保留最近一年的数据。总结:1、分区是为了分区内数据查询时扫描的数据量减少,提高查询效率2、更新模型比组件模型更适合实时数据的写入,使用更新模型后IO和CPU使用都会有明显的下降。3、对应上述第三项中会提高查询效率,字符的扫描没有数值
- 人工智能之数学基础:矩阵的范数
每天五分钟玩转人工智能
机器学习深度学习之数学基础人工智能矩阵算法线性代数范数
本文重点在前面课程中,我们学习了向量的范数,在矩阵中也有范数,本文来学习一下。矩阵的范数对于分析线性映射函数的特性有重要的作用。矩阵范数的本质矩阵范数是一种映射,它将一个矩阵映射到一个非负实数。矩阵的范数前面我们学习了向量的范数,只有当满足几个条件的时候,此时才可以,那么矩阵也是一样的,当满足下面的条件的时候,才可以定义||A||为矩阵A的范数矩阵范数的性质连续性矩阵范数是连续的函数。即如果矩阵序
- 西门子PLC S7-1200实例详解:涉及安川机器人通信、伺服电机控制及传感器数据轮询
DMQAfdLc
机器人大数据
西门子PLCS7-1200程序实例解析:电气编程者的技术之旅随着科技的飞速发展,工业自动化已成为现代制造业的核心。西门子PLC以其卓越的性能和广泛的应用,成为了工业控制领域的佼佼者。在本次技术博客中,我们将深入探讨西门子PLCS7-1200在博图版本V15下的应用实例,为电气编程者提供宝贵的学习借鉴。一、西门子PLC与安川机器人TCPIP通讯在工业自动化领域,PLC与机器人之间的通讯至关重要。西门
- python 数据可视化matplotib库安装与使用
范哥来了
信息可视化python开发语言
要使用matplotlib库进行数据可视化,首先你需要确保已经安装了该库。如果你还没有安装,可以通过Python的包管理器pip来安装它。在你的命令行工具中运行以下命令来安装matplotlib:pipinstallmatplotlib安装完成后,你就可以开始使用matplotlib来创建图表了。下面是一个简单的例子,演示如何使用matplotlib绘制一个基本的折线图。这个例子可以被添加到你当前
- HiveMetastore 的架构简析
houzhizhen
hivehive
HiveMetastore的架构简析HiveMetastore是Hive元数据管理的服务。可以把元数据存储在数据库中。对外通过api访问。hive_metastore.thrift对外提供的Thrift接口定义在文件standalone-metastore/src/main/thrift/hive_metastore.thrift中。内容包括用到的结构体和枚举,和常量,和rpcService。如分
- TCP 客户端 - 服务器通信程序搭建
Oracle_666
网络服务器tcp/ip
一、概述本文档针对TCP客户端程序和TCP服务器程序。客户端程序会连接到服务器并发送带有自定义协议格式的数据,而服务器程序则负责监听客户端连接,接收并处理这些数据。自定义协议格式为:先发送2字节网络字节序的长度头,随后是变长的数据负载。二、客户端程序2.1代码结构#include#include#include#include#include#include#definePORT8080//定义服
- FlinkCDC实战:将 MySQL 数据同步至 ES
小DuDu
flinkmysql
当前需要处理的业务场景:将订单表和相关联的表(比如:商品表、子订单表、物流信息表)组织成宽表,放入到ES中,加速订单数据的查询.同步数据到es.概述1.什么是CDC2.什么是FlinkCDC3.FlinkCDCConnectors和Flink的版本映射实战1.宽表查询1.1创建mysql表1.2启动Flink集群和FlinkSQLCLI1.3在FlinkSQLCLI中使用FlinkDDL创建表1.
- NLU-预训练模型-2018:Bert(二)【“Masked LM”缺点:①预训练与微调不一致;②忽略了掩码位置间的依赖关系】【复杂度:O(n^2·d);n:输入序列长度(规定最长512)】
u013250861
#NLP/词向量_预训练模型bert人工智能深度学习
五、BERT中的词嵌入1、为什么要使用BERT的嵌入使用BERT从文本数据中提取特征,即单词和句子的嵌入向量。我们可以用这些词和句子的嵌入向量做什么?首先,这些嵌入对于关键字/搜索扩展、语义搜索和信息检索非常有用。例如,如果你希望将客户的问题或搜索与已经回答的问题或文档化的搜索相匹配,这些表示将帮助准确的检索匹配客户意图和上下文含义的结果,即使没有关键字或短语重叠。其次,或许更重要的是,这些向量被
- 洛谷每日1题-------Day25__P1424 小鱼的航程(改进版)
__雨夜星辰__
洛谷每日1题算法c++数据结构学习笔记
题目描述有一只小鱼,它平日每天游泳250公里,周末休息(实行双休日),假设从周x开始算起,过了n天以后,小鱼一共累计游泳了多少公里呢?输入格式输入两个正整数x,n,表示从周x算起,经过n天。输出格式输出一个整数,表示小鱼累计游泳了多少公里。输入输出样例输入#1复制310输出#1复制2000说明/提示数据保证,1≤x≤7,1≤n≤106。题解#includeusingnamespacestd;int
- MATLAB中使用fread读取二进制数据时的大端序与小端序处理
知行合一←_←
matlab知识matlab开发语言
matlab里读取二进制数据时,默认按照小端序读取,怎么按照大端序读取文章目录前言一、大端序和小端序是什么?二、实际例子1.数据文件2.fread的参数总结前言只是记录matlab使用的小知识一、大端序和小端序是什么?大端序和小端序是在多个字节存储时,指定多字节数据在内存中的存储顺序,存储顺序不同,表示的值也就不同。大端序是指高位在地址较小的位置。小端序是指高位在地址较大的位置。比如地址从左到右依
- 单链表的操作
知行合一←_←
数据结构数据结构
单链表单链表是什么单链表是一种线性的链式存储结构,由多个节点组成(头结点,中间节点和尾结点),单链表的存储结构图如下:来源于网页单链表的节点是分散的,与数组不同,数组的存储结构是连续的,单链表的每个节点存储了本节点的数据和下一个节点的地址,只能单向的查找。单链表的操作单链表的操作主要包括,创建,增删改查,翻转,排序。单链表的创建单链表的创建就是创建一个头结点这里有两种创建方式,一种是仅仅创建一个头
- python 数据可视化TVTK库安装与使用
范哥来了
信息可视化python开发语言
TVTK(Traits-basedVisualizationToolKit)是一个基于Python的可视化库,它为VTK(VisualizationToolkit)提供了一个更易于使用的接口。VTK本身是非常强大的可视化工具,但使用起来可能稍微复杂一些,而TVTK通过简化API来提高易用性。下面我将指导您如何安装TVTK以及一个简单的示例来展示其基本用法。安装TVTKTVTK可以通过pip轻松安装
- 大模型微调
归一码字
人工智能
文章目录前言一、使用的库二、数据预处理1.引入库2.读入数据3.对数据进行预处理4.转换为json格式文件三,使用算子分析数据并进行数据处理四,划分训练集和测试集五,编写训练脚本开始训练六,进行模型推理人工评估总结前言这是使用知乎评论进行模型微调,让模型输出更加通畅接近人的使用语言一、使用的库modelscope:提供模型、数据集下载能力data-juicer:提供数据集处理能力ms-swift:
- 从0到1:小白也能轻松上手的高清电影搜索引擎网站制作指南
计算机学长
网站制作搜索引擎前端服务器
引言在互联网飞速发展的当下,在线观影已成为人们日常娱乐不可或缺的一部分。据相关数据显示,2024年网络视频用户规模达到了惊人的规模,如此庞大的用户群体,对电影资源的需求自然也是水涨船高。然而,面对海量的电影资源,如何快速、准确地找到自己心仪的高清电影,却成了许多影迷的一大难题。各大视频平台资源分散,想要观看不同的电影,往往需要在多个平台之间来回切换,而且还可能面临付费门槛、广告干扰等问题。这时,一
- 基于Wasm的边缘计算Pandas:突破端侧AI的最后一公里——让数据分析在手机、IoT设备上飞驰
Eqwaak00
Pandas人工智能wasm边缘计算pandas架构深度学习
引言:边缘计算的算力觉醒在智能家居设备每秒产生数万条传感器数据、手机App需要实时分析用户行为的今天,传统云计算模式面临高延迟、隐私风险、带宽成本三大挑战。本文将揭示如何通过WebAssembly(Wasm)+Pandas的技术组合,在边缘设备上实现零云端依赖的实时数据分析,并通过智慧工厂设备预测性维护案例,展示从理论到工程的全链路实现。一、技术架构设计1.1边缘计算范式演进mermaid:gra
- Spring的注解积累
yijiesuifeng
spring注解
用注解来向Spring容器注册Bean。
需要在applicationContext.xml中注册:
<context:component-scan base-package=”pagkage1[,pagkage2,…,pagkageN]”/>。
如:在base-package指明一个包
<context:component-sc
- 传感器
百合不是茶
android传感器
android传感器的作用主要就是来获取数据,根据得到的数据来触发某种事件
下面就以重力传感器为例;
1,在onCreate中获得传感器服务
private SensorManager sm;// 获得系统的服务
private Sensor sensor;// 创建传感器实例
@Override
protected void
- [光磁与探测]金吕玉衣的意义
comsci
这是一个古代人的秘密:现在告诉大家
信不信由你们:
穿上金律玉衣的人,如果处于灵魂出窍的状态,可以飞到宇宙中去看星星
这就是为什么古代
- 精简的反序打印某个数
沐刃青蛟
打印
以前看到一些让求反序打印某个数的程序。
比如:输入123,输出321。
记得以前是告诉你是几位数的,当时就抓耳挠腮,完全没有思路。
似乎最后是用到%和/方法解决的。
而今突然想到一个简短的方法,就可以实现任意位数的反序打印(但是如果是首位数或者尾位数为0时就没有打印出来了)
代码如下:
long num, num1=0;
- PHP:6种方法获取文件的扩展名
IT独行者
PHP扩展名
PHP:6种方法获取文件的扩展名
1、字符串查找和截取的方法
1
$extension
=
substr
(
strrchr
(
$file
,
'.'
), 1);
2、字符串查找和截取的方法二
1
$extension
=
substr
- 面试111
文强chu
面试
1事务隔离级别有那些 ,事务特性是什么(问到一次)
2 spring aop 如何管理事务的,如何实现的。动态代理如何实现,jdk怎么实现动态代理的,ioc是怎么实现的,spring是单例还是多例,有那些初始化bean的方式,各有什么区别(经常问)
3 struts默认提供了那些拦截器 (一次)
4 过滤器和拦截器的区别 (频率也挺高)
5 final,finally final
- XML的四种解析方式
小桔子
domjdomdom4jsax
在平时工作中,难免会遇到把 XML 作为数据存储格式。面对目前种类繁多的解决方案,哪个最适合我们呢?在这篇文章中,我对这四种主流方案做一个不完全评测,仅仅针对遍历 XML 这块来测试,因为遍历 XML 是工作中使用最多的(至少我认为)。 预 备 测试环境: AMD 毒龙1.4G OC 1.5G、256M DDR333、Windows2000 Server
- wordpress中常见的操作
aichenglong
中文注册wordpress移除菜单
1 wordpress中使用中文名注册解决办法
1)使用插件
2)修改wp源代码
进入到wp-include/formatting.php文件中找到
function sanitize_user( $username, $strict = false
- 小飞飞学管理-1
alafqq
管理
项目管理的下午题,其实就在提出问题(挑刺),分析问题,解决问题。
今天我随意看下10年上半年的第一题。主要就是项目经理的提拨和培养。
结合我自己经历写下心得
对于公司选拔和培养项目经理的制度有什么毛病呢?
1,公司考察,选拔项目经理,只关注技术能力,而很少或没有关注管理方面的经验,能力。
2,公司对项目经理缺乏必要的项目管理知识和技能方面的培训。
3,公司对项目经理的工作缺乏进行指
- IO输入输出部分探讨
百合不是茶
IO
//文件处理 在处理文件输入输出时要引入java.IO这个包;
/*
1,运用File类对文件目录和属性进行操作
2,理解流,理解输入输出流的概念
3,使用字节/符流对文件进行读/写操作
4,了解标准的I/O
5,了解对象序列化
*/
//1,运用File类对文件目录和属性进行操作
//在工程中线创建一个text.txt
- getElementById的用法
bijian1013
element
getElementById是通过Id来设置/返回HTML标签的属性及调用其事件与方法。用这个方法基本上可以控制页面所有标签,条件很简单,就是给每个标签分配一个ID号。
返回具有指定ID属性值的第一个对象的一个引用。
语法:
&n
- 励志经典语录
bijian1013
励志人生
经典语录1:
哈佛有一个著名的理论:人的差别在于业余时间,而一个人的命运决定于晚上8点到10点之间。每晚抽出2个小时的时间用来阅读、进修、思考或参加有意的演讲、讨论,你会发现,你的人生正在发生改变,坚持数年之后,成功会向你招手。不要每天抱着QQ/MSN/游戏/电影/肥皂剧……奋斗到12点都舍不得休息,看就看一些励志的影视或者文章,不要当作消遣;学会思考人生,学会感悟人生
- [MongoDB学习笔记三]MongoDB分片
bit1129
mongodb
MongoDB的副本集(Replica Set)一方面解决了数据的备份和数据的可靠性问题,另一方面也提升了数据的读写性能。MongoDB分片(Sharding)则解决了数据的扩容问题,MongoDB作为云计算时代的分布式数据库,大容量数据存储,高效并发的数据存取,自动容错等是MongoDB的关键指标。
本篇介绍MongoDB的切片(Sharding)
1.何时需要分片
&nbs
- 【Spark八十三】BlockManager在Spark中的使用场景
bit1129
manager
1. Broadcast变量的存储,在HttpBroadcast类中可以知道
2. RDD通过CacheManager存储RDD中的数据,CacheManager也是通过BlockManager进行存储的
3. ShuffleMapTask得到的结果数据,是通过FileShuffleBlockManager进行管理的,而FileShuffleBlockManager最终也是使用BlockMan
- yum方式部署zabbix
ronin47
yum方式部署zabbix
安装网络yum库#rpm -ivh http://repo.zabbix.com/zabbix/2.4/rhel/6/x86_64/zabbix-release-2.4-1.el6.noarch.rpm 通过yum装mysql和zabbix调用的插件还有agent代理#yum install zabbix-server-mysql zabbix-web-mysql mysql-
- Hibernate4和MySQL5.5自动创建表失败问题解决方法
byalias
J2EEHibernate4
今天初学Hibernate4,了解了使用Hibernate的过程。大体分为4个步骤:
①创建hibernate.cfg.xml文件
②创建持久化对象
③创建*.hbm.xml映射文件
④编写hibernate相应代码
在第四步中,进行了单元测试,测试预期结果是hibernate自动帮助在数据库中创建数据表,结果JUnit单元测试没有问题,在控制台打印了创建数据表的SQL语句,但在数据库中
- Netty源码学习-FrameDecoder
bylijinnan
javanetty
Netty 3.x的user guide里FrameDecoder的例子,有几个疑问:
1.文档说:FrameDecoder calls decode method with an internally maintained cumulative buffer whenever new data is received.
为什么每次有新数据到达时,都会调用decode方法?
2.Dec
- SQL行列转换方法
chicony
行列转换
create table tb(终端名称 varchar(10) , CEI分值 varchar(10) , 终端数量 int)
insert into tb values('三星' , '0-5' , 74)
insert into tb values('三星' , '10-15' , 83)
insert into tb values('苹果' , '0-5' , 93)
- 中文编码测试
ctrain
编码
循环打印转换编码
String[] codes = {
"iso-8859-1",
"utf-8",
"gbk",
"unicode"
};
for (int i = 0; i < codes.length; i++) {
for (int j
- hive 客户端查询报堆内存溢出解决方法
daizj
hive堆内存溢出
hive> select * from t_test where ds=20150323 limit 2;
OK
Exception in thread "main" java.lang.OutOfMemoryError: Java heap space
问题原因: hive堆内存默认为256M
这个问题的解决方法为:
修改/us
- 人有多大懒,才有多大闲 (评论『卓有成效的程序员』)
dcj3sjt126com
程序员
卓有成效的程序员给我的震撼很大,程序员作为特殊的群体,有的人可以这么懒, 懒到事情都交给机器去做 ,而有的人又可以那么勤奋,每天都孜孜不倦得做着重复单调的工作。
在看这本书之前,我属于勤奋的人,而看完这本书以后,我要努力变成懒惰的人。
不要在去庞大的开始菜单里面一项一项搜索自己的应用程序,也不要在自己的桌面上放置眼花缭乱的快捷图标
- Eclipse简单有用的配置
dcj3sjt126com
eclipse
1、显示行号 Window -- Prefences -- General -- Editors -- Text Editors -- show line numbers
2、代码提示字符 Window ->Perferences,并依次展开 Java -> Editor -> Content Assist,最下面一栏 auto-Activation
- 在tomcat上面安装solr4.8.0全过程
eksliang
Solrsolr4.0后的版本安装solr4.8.0安装
转载请出自出处:
http://eksliang.iteye.com/blog/2096478
首先solr是一个基于java的web的应用,所以安装solr之前必须先安装JDK和tomcat,我这里就先省略安装tomcat和jdk了
第一步:当然是下载去官网上下载最新的solr版本,下载地址
- Android APP通用型拒绝服务、漏洞分析报告
gg163
漏洞androidAPP分析
点评:记得曾经有段时间很多SRC平台被刷了大量APP本地拒绝服务漏洞,移动安全团队爱内测(ineice.com)发现了一个安卓客户端的通用型拒绝服务漏洞,来看看他们的详细分析吧。
0xr0ot和Xbalien交流所有可能导致应用拒绝服务的异常类型时,发现了一处通用的本地拒绝服务漏洞。该通用型本地拒绝服务可以造成大面积的app拒绝服务。
针对序列化对象而出现的拒绝服务主要
- HoverTree项目已经实现分层
hvt
编程.netWebC#ASP.ENT
HoverTree项目已经初步实现分层,源代码已经上传到 http://hovertree.codeplex.com请到SOURCE CODE查看。在本地用SQL Server 2008 数据库测试成功。数据库和表请参考:http://keleyi.com/a/bjae/ue6stb42.htmHoverTree是一个ASP.NET 开源项目,希望对你学习ASP.NET或者C#语言有帮助,如果你对
- Google Maps API v3: Remove Markers 移除标记
天梯梦
google maps api
Simply do the following:
I. Declare a global variable:
var markersArray = [];
II. Define a function:
function clearOverlays() {
for (var i = 0; i < markersArray.length; i++ )
- jQuery选择器总结
lq38366
jquery选择器
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40
- 基础数据结构和算法六:Quick sort
sunwinner
AlgorithmQuicksort
Quick sort is probably used more widely than any other. It is popular because it is not difficult to implement, works well for a variety of different kinds of input data, and is substantially faster t
- 如何让Flash不遮挡HTML div元素的技巧_HTML/Xhtml_网页制作
刘星宇
htmlWeb
今天在写一个flash广告代码的时候,因为flash自带的链接,容易被当成弹出广告,所以做了一个div层放到flash上面,这样链接都是a触发的不会被拦截,但发现flash一直处于div层上面,原来flash需要加个参数才可以。
让flash置于DIV层之下的方法,让flash不挡住飘浮层或下拉菜单,让Flash不档住浮动对象或层的关键参数:wmode=opaque。
方法如下:
- Mybatis实用Mapper SQL汇总示例
wdmcygah
sqlmysqlmybatis实用
Mybatis作为一个非常好用的持久层框架,相关资料真的是少得可怜,所幸的是官方文档还算详细。本博文主要列举一些个人感觉比较常用的场景及相应的Mapper SQL写法,希望能够对大家有所帮助。
不少持久层框架对动态SQL的支持不足,在SQL需要动态拼接时非常苦恼,而Mybatis很好地解决了这个问题,算是框架的一大亮点。对于常见的场景,例如:批量插入/更新/删除,模糊查询,多条件查询,联表查询,
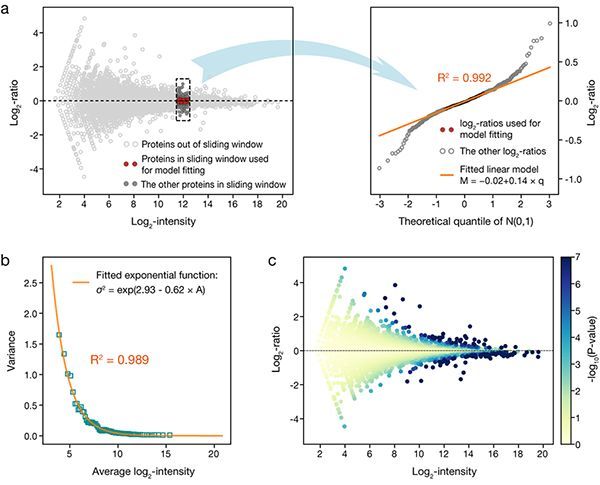 图一:MAP模型的分步回归分析流程:(a)局域线性拟合;(b)全局指数拟合构建技术误差模型;(c)计算每个蛋白质信号差异的显著性P值。
图一:MAP模型的分步回归分析流程:(a)局域线性拟合;(b)全局指数拟合构建技术误差模型;(c)计算每个蛋白质信号差异的显著性P值。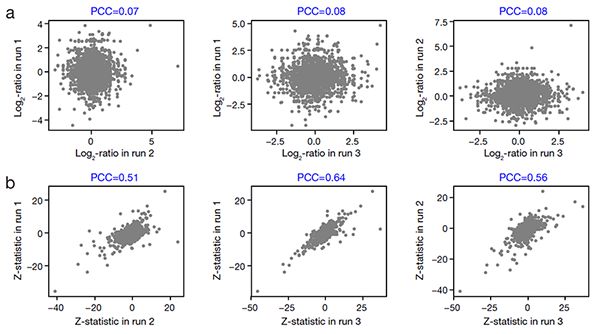 图二:不同批次蛋白质iTRAQ信号的log2比率(a)和Z统计量(b)的皮尔森关联系数。
图二:不同批次蛋白质iTRAQ信号的log2比率(a)和Z统计量(b)的皮尔森关联系数。