生信&数据挖掘——人工神经网络篇(3)差异分析
在这篇文章中,我将讨论如何使用基因表达差异分析(DEA)来进行差异分析。DEA是一种有效的方法,可以从大量基因表达数据中检测和预测差异。我将介绍DEA的基本原理,并讨论如何使用它来进行差异分析。我还将介绍一些常用的DEA技术,以及它们如何用于差异分析。最后,我将介绍一些实际应用,以及如何使用DEA来解决实际问题。
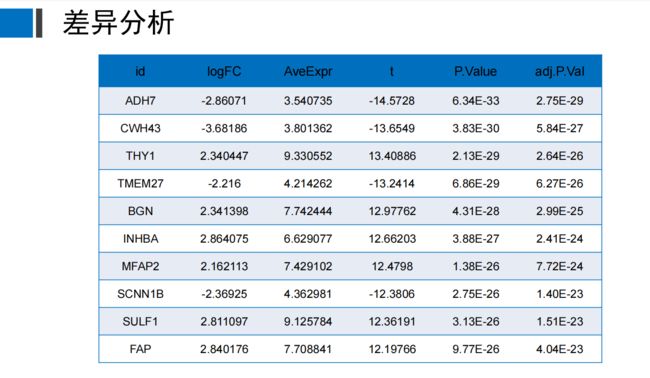
先看看最终结果和名词解释
ID:ID是一个唯一的标识符,用于标识每个样本。
logFC:logFC是对数变化率的缩写,它表示两个样本之间的变化率,以对数形式表示。 (大于1上调,小于1下调)也可以是2(看今天需求)
AveExpr:AveExpr是平均表达量的缩写,它表示每个样本的平均表达量。
P.Value:P.Value是P值的缩写,它表示两个样本之间的差异是否显著。
P < 0.05:表示统计检验的结果显示,观察到的结果是显著的,概率值小于0.05,表明结果是有统计学意义的。
P 0.01:表示统计检验的结果显示,观察到的结果是极其显著的,概率值小于0.01,表明结果是有极强的统计学意义的。
P ≤ 0.001:表示统计检验的结果显示,观察到的结果是极其显著的,概率值小于或等于0.001,表明结果是有极强的统计学意义的。
P 0.0001:表示统计检验的结果显示,观察到的结果是极其显著的,概率值小于0.0001,表明结果是有极强的统计学意义的。
t:检验值,用于检验两组样本之间的差异是否具有统计学意义。
adj.P.Val:adj.P.Val是调整后的P值的缩写,它表示两个样本之间的差异是否显著,考虑了多重比较的影响。(一般看p值不看调整后的P值)
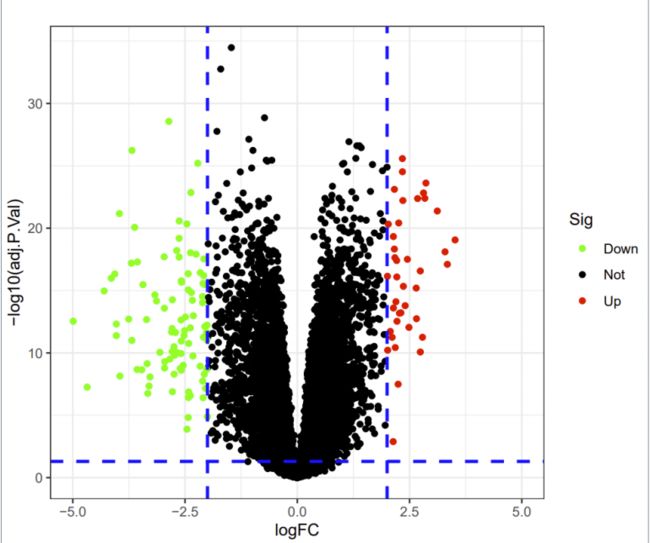
差异基因火山图
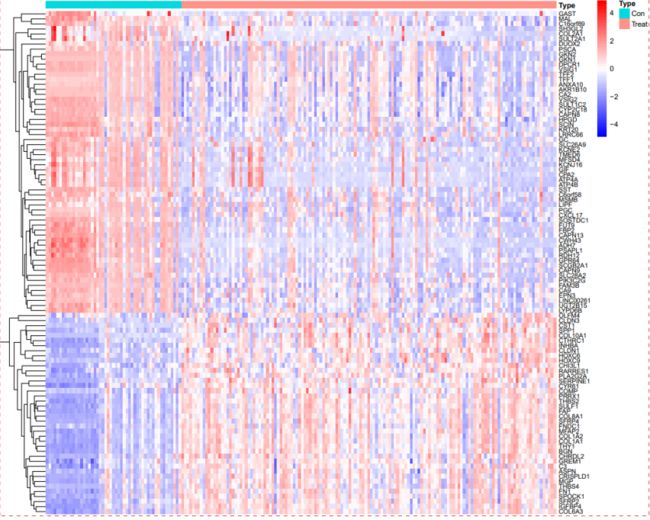
一般我们用于显示基因表达水平之间的差异。它通过将基因表达水平的差异映射到一个热图上,以便更容易地比较和观察不同样本之间的差异。热图中的右侧一行代表一个基因,颜色代表基因表达水平的差异,红色表示表达水平较高(高表达),蓝色表示表达水平较低(低表达)。
GSE65801和GSE54129数据先合并一下
输入文件:注释好的基因表达数据,只需要将txt放在和脚本同一文件夹里面

#引用包
library(limma)
library(sva)
outFile="merge.txt" #输出文件
setwd("C:\\05.sva") #设置工作目录
#获取目录下所有".txt"结尾的文件
files=dir()
files=grep("txt$", files, value=T)
geneList=list()
#读取所有txt文件中的基因信息,保存到geneList
for(file in files){
if(file==outFile){next}
rt=read.table(file, header=T, sep="\t", check.names=F) #读取输入文件
geneNames=as.vector(rt[,1]) #提取基因名称
uniqGene=unique(geneNames) #基因取unique
header=unlist(strsplit(file, "\\.|\\-"))
geneList[[header[1]]]=uniqGene
}
#获取交集基因
interGenes=Reduce(intersect, geneList)
#数据合并
allTab=data.frame()
batchType=c()
for(i in 1:length(files)){
inputFile=files[i]
header=unlist(strsplit(inputFile, "\\.|\\-"))
#读取输入文件,并对输入文件进行整理
rt=read.table(inputFile, header=T, sep="\t", check.names=F)
rt=as.matrix(rt)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
rt=avereps(data)
#对数值大的数据取log2
qx=as.numeric(quantile(rt, c(0, 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))
LogC=( (qx[5]>100) || ( (qx[6]-qx[1])>50 && qx[2]>0) )
if(LogC){
rt[rt<0]=0
rt=log2(rt+1)}
rt=normalizeBetweenArrays(rt)
#数据合并
if(i==1){
allTab=rt[interGenes,]
}else{
allTab=cbind(allTab, rt[interGenes,])
}
batchType=c(batchType, rep(i,ncol(rt)))
}
#对数据进行矫正,输出矫正后的结果
outTab=ComBat(allTab, batchType, par.prior=TRUE)
outTab=rbind(geneNames=colnames(outTab), outTab)
write.table(outTab, file="merge.txt", sep="\t", quote=F, col.names=F)结果生成merge.txt

将merge.txt和他们对照组和实验组的信息放在同一个文件夹

脚本如下:
#if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#BiocManager::install("limma")
#install.packages("pheatmap")
#引用包
library(limma)
library(pheatmap)
inputFile="merge.txt" #输入文件
logFCfilter=2 #logFC过滤阈值
adj.P.Val.Filter=0.05 #矫正后p值阈值
setwd("C:\\biowolf\\Diagnostic\\06.diff") #设置工作目录
#读取输入文件,并对输入文件整理
rt=read.table(inputFile, header=T, sep="\t", check.names=F)
rt=as.matrix(rt)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
data=avereps(data)
data=data[rowMeans(data)>0,]
#读取目录下所有"s1.txt"结尾的文件
sampleName1=c()
files=dir()
files=grep("s1.txt$", files, value=T)
for(file in files){
rt=read.table(file, header=F, sep="\t", check.names=F) #读取输入文件
geneNames=as.vector(rt[,1]) #提取基因名称
uniqGene=unique(geneNames) #基因取unique
sampleName1=c(sampleName1, uniqGene)
}
#读取目录下所有"s2.txt"结尾的文件
sampleName2=c()
files=dir()
files=grep("s2.txt$", files, value=T)
for(file in files){
rt=read.table(file, header=F, sep="\t", check.names=F) #读取输入文件
geneNames=as.vector(rt[,1]) #提取基因名称
uniqGene=unique(geneNames) #基因取unique
sampleName2=c(sampleName2, uniqGene)
}
#提取实验组和对照组的数据
conData=data[,sampleName1]
treatData=data[,sampleName2]
data=cbind(conData,treatData)
conNum=ncol(conData)
treatNum=ncol(treatData)
#差异分析
Type=c(rep("con",conNum),rep("treat",treatNum))
design <- model.matrix(~0+factor(Type))
colnames(design) <- c("con","treat")
fit <- lmFit(data,design)
cont.matrix<-makeContrasts(treat-con,levels=design)
fit2 <- contrasts.fit(fit, cont.matrix)
fit2 <- eBayes(fit2)
allDiff=topTable(fit2,adjust='fdr',number=200000)
allDiffOut=rbind(id=colnames(allDiff),allDiff)
write.table(allDiffOut, file="all.txt", sep="\t", quote=F, col.names=F)
#输出矫正后的表达量
outData=rbind(id=paste0(colnames(data),"_",Type),data)
write.table(outData, file="normalize.txt", sep="\t", quote=F, col.names=F)
#输出差异结果
diffSig=allDiff[with(allDiff, (abs(logFC)>logFCfilter & adj.P.Val < adj.P.Val.Filter )), ]
diffSigOut=rbind(id=colnames(diffSig),diffSig)
write.table(diffSigOut, file="diff.txt", sep="\t", quote=F, col.names=F)
#输出差异基因表达量
diffGeneExp=data[row.names(diffSig),]
diffGeneExpOut=rbind(id=paste0(colnames(diffGeneExp),"_",Type), diffGeneExp)
write.table(diffGeneExpOut, file="diffGeneExp.txt", sep="\t", quote=F, col.names=F)
#绘制差异基因热图
geneNum=50
diffSig=diffSig[order(as.numeric(as.vector(diffSig$logFC))),]
diffGeneName=as.vector(rownames(diffSig))
diffLength=length(diffGeneName)
hmGene=c()
if(diffLength>(2*geneNum)){
hmGene=diffGeneName[c(1:geneNum,(diffLength-geneNum+1):diffLength)]
}else{
hmGene=diffGeneName
}
hmExp=data[hmGene,]
Type=c(rep("Con",conNum),rep("Treat",treatNum))
names(Type)=colnames(data)
Type=as.data.frame(Type)
pdf(file="heatmap.pdf", width=10, height=8)
pheatmap(hmExp,
annotation=Type,
color = colorRampPalette(c("blue", "white", "red"))(50),
cluster_cols =F,
show_colnames = F,
scale="row",
fontsize = 8,
fontsize_row=7,
fontsize_col=8)
dev.off()最终输出热图和差异表达的值
绘制火山图
输入文件 all.txt 上一步差异基因图得到的
脚本如下:
#install.packages("ggplot2")
library(ggplot2) #引用包
logFCfilter=2 #logFC过滤条件
adj.P.Val.Filter=0.05 #矫正后的p值过滤条件
inputFile="all.txt" #输入文件
setwd("C:\\biowolf\\neuralDiagnostic\\07.volcano") #设置工作目录
#读取输入文件
rt=read.table(inputFile, header=T, sep="\t", check.names=F)
#定义显著性
Sig=ifelse((rt$adj.P.VallogFCfilter), ifelse(rt$logFC>logFCfilter,"Up","Down"), "Not")
rt=cbind(rt, Sig=Sig)
#绘制火山图
p=ggplot(rt, aes(logFC, -log10(adj.P.Val)))+
geom_point(aes(col=Sig))+
scale_color_manual(values=c("green", "black", "red"))+
xlim(-5,5)+
labs(title = " ")+
geom_vline(xintercept=c(-logFCfilter,logFCfilter), col="blue", cex=1, linetype=2)+
geom_hline(yintercept= -log10(adj.P.Val.Filter), col="blue", cex=1, linetype=2)+
theme(plot.title=element_text(size=16, hjust=0.5, face="bold"))
p=p+theme_bw()
#输出火山图
pdf(file="volcano.pdf", width=6, height=5.1)
print(p)
dev.off()
绿色是下调,红色是上调的基因