教程 | 如何用cd-hit去除冗余序列?
0.简介
生信分析中经常要根据指定条件查找相似序列,比如构建多个样品间的非冗余基因集、分析样品间的相似程度等等,cd-hit这款软件就可以用较短的时间解决此类问题,可以对单个数据集进行去冗余,包括DNA/RNA序列和蛋白序列,也可以对两个数据集进行比较。其工作原理可概述为:将所有序列按照参数设定进行聚类,并将每一组聚类中的最长序列作为代表序列进行输出,同时给出每组聚类下的每个序列名可供相似度分析使用。下面我们来简单介绍一下它的使用方法。
1.下载与安装
网址:http://cd-hit.org ;http://www.bioinformatics.org/cd-hit/ ;https://github.com/weizhongli/cdhit/archive/V4.6.2.tar.gz;
这是一个在linux系统下使用的工作,我们可以给自己的电脑装一个双系统或者在windows下使用linux的虚拟机。然后我们可以执行下面的命令进行解压(注意我们要将路径先切换到安装包所在的文件目录下,或者在执行命令时使用完整路径)。
gzip -d cdhit-4.2.tar.gz然后进入到解压后的文件夹(我解压后的文件夹为cdhit-4.2,同样要注意我们的文件路径问题,如果上面使用的是完整路径,最好这里也使用完整路径,比如我使用完整路径是‘cd /home/zpf/cdhit-4.2’)
cd cdhit-4.2最后编译一下就可以了,执行make
make然后我们就可以使用这个工具了。
2.输入文件格式
Cd-hit的输入文件仅有一个fasta格式文件 ,一般来说cd-hit是将几个样品的基因或蛋白序列进行聚类,所以需要将这些样品的序列汇总到一起作为输入文件,可在linux系统下通过cat命令实现:
cat a.fasta b.fasta c.fasta > all.fasta其中a.fasta,b.fasta,c.fasta为fasta格式的三个样品基因或蛋白序列,all.fasta为汇总后的序列,在分析中作为cd-hit的输入序列。值得注意的是,在三个样品序列中不能有序列名相同的序列,否则会出现错误。因此,一般在分析时会在各样品序列名前添加样品名,这样即可避免重复。序列名是fasta文件中以“>”开头的行空格之前的内容,如下图中蓝色线圈出部分。
 图1
图1
3.输出文件
Cd-hit有两个输出文件:一个是只含有所有代表序列(即去冗余后的序列)的fasta文件,其格式参看图1;另一个是以.clstr结尾的聚类信息文件,其格式如图2。
 图2
图2
以“>”开头的是一个聚类组。每组下面按序号排列,如上图中Cluster 1组有5个聚类序列。每个聚类序列有一个百分比或“*”,百分比代表该序列与代表序列的相似度,“*”代表该序列即为代表序列。
4.去除冗余的基本思路
首先对所有序列按照其长度进行排序,然后从最长的序列开始,形成第一个序列类,然后依次对序列进行处理,如果新的序列与已有的序列类的代表序列的相似性在cutoff以上则把该序列加到该序列类中,否则形成新的序列类。之所以快主要是两个方面的原因:一个是使用了word过滤方法,即如果两条序列之间的相似性在80%(假设序列长度为100),那么它们至少有60个相同的长度为2的word,至少有40个相同的长度为3的word,至少有20个相同的长度为4的word。基于这个原则,在处理新的序列的时候,如果新的序列与已有序列的相同word的长度不能满足这些要求则不需要进行比对了,这极大的降低了时间消耗;另外一个速度快的原因是使用了index table,可以很快的计算序列之间相同word的数目。
#当序列相似性在80%时,有20个位点是有差异的,极端的情况就是这20个位点对应的长度为2的字符串都不一样,因此是40个不一样,当有更多的不一样时,两条序列的相似性不可能在80%;同理,如果这20个位点对应的长度为4的字符串都不一样,则有80个不一样。
5.使用方法和参数介绍
Cd-hit运行时用很多参数可以进行调整设置,其运行命令为(参数仅为示例)在刚才编译的文件路径下执行:
cd-hit -i all.fasta -o new.fa -c 0.8 -aS 0.8 -d 0下面简单介绍一下重要的几个参数:
-i:输入文件,fasta格式。
-o:输出文件前缀,输出文件有两个,分别为fasta格式序列文件和以.clstr结尾的聚类信息文件。
-c:较短序列比对到长序列的bp与自身bp数的比值超过该数值则聚类为一组,默认为0.9。
-d:聚类信息文件中各个聚类组中序列名的长度,设为0则将取完整序列名。
-aL:控制代表序列比对严格程度的参数,默认为0,若设为0.8则表示比对区间要占到代表(长)序列的80%。
-AL:控制代表序列比对严格程度的参数,默认为99999999,若设为40则表示代表序列的非比对区间要短于40bp。
-aS:控制短序列比对严格程度的参数,默认为0,若设为0.8则表示比对区间要占到短序列的80%。
-AS:控制短序列比对严格程度的参数,默认为99999999,若设为40则表示短序列的非比对区间要短于40bp。
下图详解了-aL,-AL,-aS,-AS四个参数。
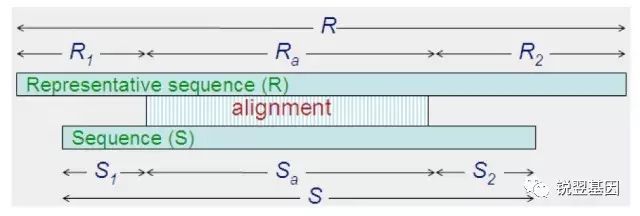 图3
图3
aL = Ra / R
AL = R - Ra
aS = Sa / S
AS = S - Sa
6.cdhit的缺点
1 它不能保证同一个序列类中的序列的相似性都在threshold之上,因为每次比对都是用新序列与序列类的代表序列进行,这就有可能使得序列类中除了代表序列外其他序列之间的相似性在threshold之下。比如A是代表序列,B与A的相似性大于0.95,C与A的相似性也大于0.95,但是这并不能保证B与C的相似性也大于0.95.
2 它不能保证一个序列类的病毒与另外一个序列类中的病毒的相似性也在threshold之上,原因还是在于用代表序列代表了整个序列类。
3 基于word filter的方法使得使用每个长度的word能够处理的冗余性水平有限,如使用长度为2的word只能够得到相似性在50%以上的序列,长度为3的word只能够得到相似性在66.7%以上的序列类,类似的,长度为5的word只能够得到相似性在80%以上的序列。在实际应用的时候需要注意选择的word长度与threshold的匹配。