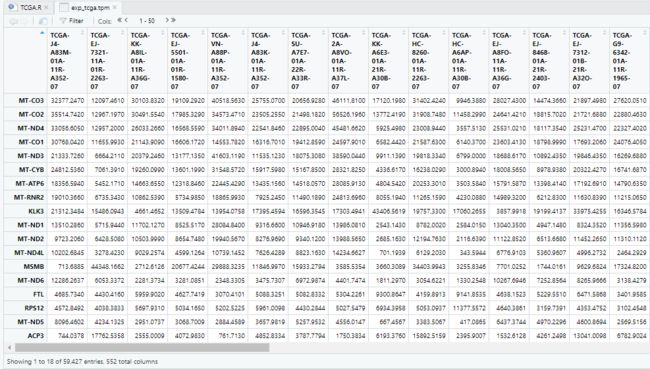
TCGA_联合GTEx分析1_得到表达矩阵.tpm
GTEx数据库获取表达矩阵.tpm
一、下载数据
共要下载三个数据,分别为表达矩阵、样本信息、注释信息
进入网站:UCSC Xena
点击“Launch Xena”,选择“DATA SETs”

点击“GTEX(11 datasets)”
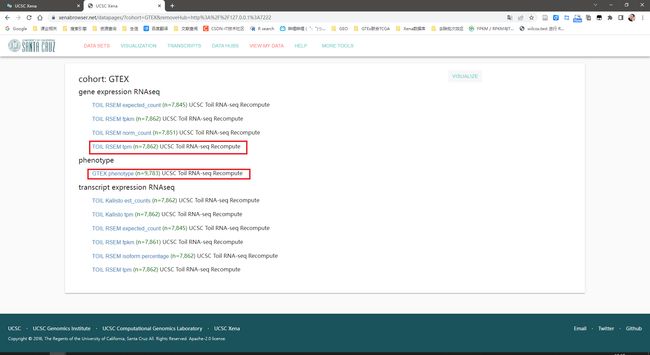
下载框中的两个数据,上面一个是表达矩阵,下面一个是样本信息。还差一个注释信息,下载地址:https://toil.xenahubs.net/download/probeMap/gencode.v23.annotation.gene.probemap
需要注意的是:
表达矩阵中数据格式为log2(tpm+0.001)
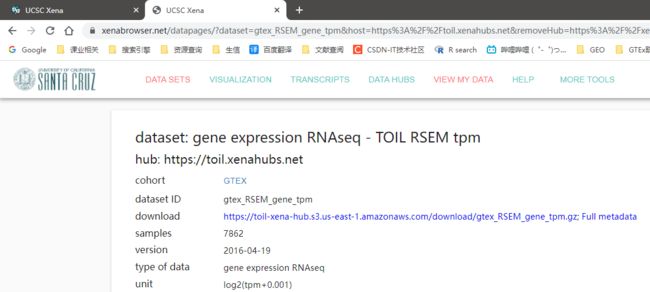
下载完成后,三个文件的文件名分别为:
- gtex_RSEM_gene_tpm.gz
- GTEX_phenotype.gz
- gencode.v23.annotation.gene.probemap
二、载入数据
library(data.table) #载入数据用#表达矩阵
exp_gtex.tpm=fread("gtex_RSEM_gene_tpm.gz",header = T, sep = '\t',data.table = F)
rownames(exp_gtex.tpm)=exp_gtex.tpm[,1]
exp_gtex.tpm=exp_gtex.tpm[,-1]
#样本信息
data_cl=fread("GTEX_phenotype.gz",header = T, sep = '\t',data.table = F)
data_cl=data_cl[,c(1,3)]
names(data_cl)=c('Barcode','Tissue')
data_cl=data_cl[data_cl$Tissue == 'Prostate',] #筛选出Prostate的数据
#注释信息
annotat=fread("gencode.v23.annotation.gene.probemap",header = T, sep = '\t',data.table = F)
annotat=annotat[,c(1,2)]
rownames(annotat)=annotat[,1] #这里没有选择删去id这一列View(exp_gtex.tpm)
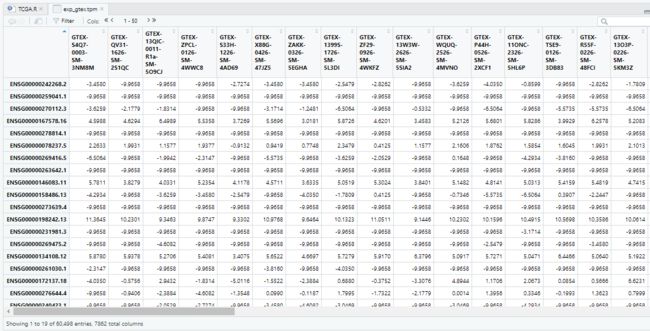
View(data_cl)
样本信息中有122个barcode来自Prostate组织

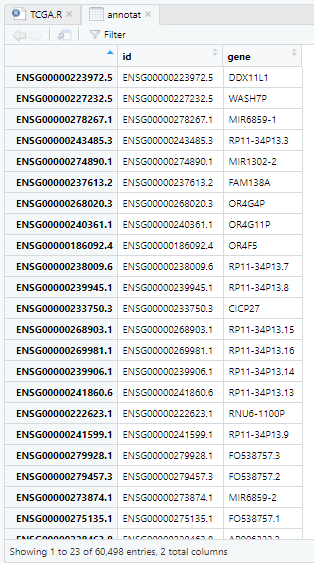
View(annotat)
三、处理数据
1 筛选出exp_gtex.tpm中的Prostate组织数据,并还原为TPM
#筛选,筛选之后还剩100个barcode
exp_gtex.tpm=exp_gtex.tpm[,colnames(exp_gtex.tpm) %in% data_cl$Barcode]
#还原为TPM
exp_gtex.tpm=2^exp_gtex.tpm-0.0012 基因注释,去重复基因名,读出表达矩阵
#基因注释
exp_gtex.tpm=as.matrix(exp_gtex.tpm)
t_index=intersect(rownames(exp_gtex.tpm),rownames(annotat)) #行名取交集,t_index中是能够进行注释的probe_id
exp_gtex.tpm=exp_gtex.tpm[t_index,]
annotat=annotat[t_index,]
rownames(exp_gtex.tpm)=annotat$gene
#去除重复基因名
t_index1=order(rowMeans(exp_gtex.tpm),decreasing = T)
t_data_order=exp_gtex.tpm[t_index1,]
keep=!duplicated(rownames(t_data_order))#对于有重复的基因,保留第一次出现的那个,即行平均值大的那个
exp_gtex.tpm=t_data_order[keep,]#得到最后处理之后的表达谱矩阵
#读出
write.csv(exp_gtex.tpm,file = "exp_gtex.tpm.csv",quote = FALSE) View(exp_gtex.tpm)

TCGA数据库获取表达矩阵.tpm
TCGA_改版后STAR-count处理方法_老实人谢耳朵的博客-CSDN博客
result <- fromJSON(file = "E:/R/PRAD Data Mining/PRAD_data_mining/TCGA/Results/DESeq2差异分析/TP vs NT/GDCdata_star_count_TP&NT/metadata.cart.2022-05-01.json")
metadata <- data.frame(t(sapply(result,function(x){
id <- x$associated_entities[[1]]$entity_submitter_id
file_name <- x$file_name
all <- cbind(id,file_name)
})))
rownames(metadata) <- metadata[,2]
#获取raw
t_dir <- 'E:/R/PRAD Data Mining/PRAD_data_mining/TCGA/Results/DESeq2差异分析/TP vs NT/GDCdata_star_count_TP&NT/all/'
t_samples=list.files(t_dir)
sampledir <- paste0(t_dir,t_samples) #各个文件路径
example <- data.table::fread('E:/R/PRAD Data Mining/PRAD_data_mining/TCGA/Results/DESeq2差异分析/TP vs NT/GDCdata_star_count_TP&NT/all/005d2b9e-722c-40bd-aa5c-bd4e8842cb04.rna_seq.augmented_star_gene_counts.tsv',data.table = F)#读入一个tsv文件,查看需要的列数,“unstranded”
raw <- do.call(cbind,lapply(sampledir, function(x){
rt <- data.table::fread(x,data.table = F) #data.table::fread函数
rownames(rt) <- rt[,1]
rt <- rt[,7]###第7列为“tpm_unstranded”
}))
#替换行名、列名
colnames(raw)=sapply(strsplit(sampledir,'/'),'[',11)###列名,11为文件名005d2b9e-722c-40bd-aa5c-bd4e8842cb04.rna_seq.augmented_star_gene_counts.tsv
rownames(raw) <- example$gene_id ##行名
raw_t <- t(raw)
t_same <- intersect(row.names(metadata),row.names(raw_t))
dataPrep2 <- cbind(metadata[t_same,],raw_t[t_same,])
rownames(dataPrep2) <- dataPrep2[,1]
dataPrep2 <- t(dataPrep2)
dataPrep2 <-dataPrep2[-c(1:6),] #dataPrep2为未注释count矩阵
#dataPrep2中数据类型为“character”,需要转为“numeric”
puried_data=apply(dataPrep2,2,as.numeric)
#基因注释
rownames(puried_data)=example[5:nrow(example),'gene_name']
#去除重复基因名
t_index=order(rowMeans(puried_data),decreasing = T)#计算所有行平均值,按降序排列
t_data_order=puried_data[t_index,]#调整表达谱的基因顺序
keep=!duplicated(rownames(t_data_order))#对于有重复的基因,保留第一次出现的那个,即行平均值大的那个
exp_tcga.tpm=t_data_order[keep,]#得到最后处理之后的表达谱矩阵
write.csv(exp_tcga.tpm,file = "exp_tcga.tpm.csv",quote = FALSE) View(exp_tcga.tpm)