CADD之分子对接一:背景介绍
在药物筛选过程中,当靶标的结构已知时,可以用计算工具虚拟筛选找到能够调控蛋白质功能的小分子,这种方法叫做基于结构的虚拟筛选(structure-based virtual screening, SBVS),目前,SBVS已成为药物发现的一种常规技术,并且被用于其他需要筛选活性化合物的研究领域。SBVS可以看成是对传统实验高通量筛选(HTS)的一种计算机模拟。
SBVS方法需要得到靶蛋白的三维(3D)结构及小分子的3D结构:
靶蛋白三维(3D)结构获取:PDB,PDBsum数据库(通过晶体学,核磁共振波谱学或同源建模等方法获得);
小分子的3D结构获取:直接在zinc库中获取3D结构,或得到smiles,mol2等格式低维文件,通过openbabel转化为pdbqt格式。
在SBVS方法中,我们将化合物"放置"到靶蛋白的指定活性位点,在活性化合物的不同姿态(构象)中找到活性构象,并进行打分,通过打分函数区分活性化合物和非活性化合物,这一过程叫做分子对接。
目前已开发出的分子对接程序有60多种,打分函数有30多种,部分程序(AutoDock、Dock、Vina、FlexX,FRED,Glide,GOLD,ICM)在计算化学界认可度很高。
熟悉了背景和工具以后,下面重点介绍分子对接的基础。
分子对接基本理论:
现阶段分子对接的基本理论模型是诱导—契合模型,包括配体和靶蛋白在两个层次上的匹配:
1.结构匹配;
结构上一般是衡量RMSD值,小于2埃米是比较好的
2.能量匹配。
通常为体系的吉布斯自由能,能量越低越稳定
分子对接中,几种常见蛋白质残基与小分子受体的相互作用:
1.金属配位作用:一些蛋白质含有金属离子作为辅助因子,金属离子与配体的给电子或官能团的配位作用通常对配体的整体结合亲和力贡献最大,也决定了配体的作用机制。
2.离子相互作用(盐桥):离子相互作用是很强的(>400KJ/mol)吸引作用,发生在配体与蛋白质的带相反电荷的基团之间(正负可电离区域可以是单个原子(如金属阳离子),也可以是能够在生理pH下被质子化基团或去质子化基团(如:羧酸,胍基,芳香杂环等))。
离子相互作用是一种静电作用,无方向性
3.氢键相互作用:氢键是形成特异性的配体—受体复合物时最重要的特异性相互作用(为了描述氢键相互作用的特征,通常将其建模为对受体(或供体)原子的位置(及位置变化的公差))。氢键一般在2.5—3.5埃米之间,3—3.5之间的氢键较弱,2.5—3之间的氢键较强。
氢键有方向约束及供体受体特征
4.疏水作用:由于疏水相互作用是无方向性的,它们可以用无无约束的特征表示,表示为位于配体疏水链,分支或基团中心的公差球。-200~-100J/mol之间。
5.芳香环与阳离子—π相互作用:由于阳离子—π和π—π相互作用需要相互作用的两个化学结构之间具有一定的相对几何构型,具有方向约束。
常见分子对接的几种类型:
1.刚性对接
不考虑参与对接的分子构象变化,在计算时仅改变分子在空间上的相对位置和取向,这种方法计算量相对较小,适合于处理大分子之间(如:Pr-Pr,Pr-核酸或核酸核酸)的对接。代表性对接程序有(Cluspro,Hex,HDOCK和ZDOCK等)
2.半柔性对接
半柔性对接需要考虑参与对接的配体分子的构象变化,在计算时不但要改变分子在空间上的相对位置和取向,同时还要对小分子的构象进行搜索,这种方法兼顾了计算量和模型预测能力,是目前主流的分子对接方法。适用于大分子和小分子的对接,(代表性对接程序有AutoDock vina,UCSFDock,Glide和GoLD等)。
3.柔性对接
需要同时考虑参与对接的受体和配体分子的构象变化,计算时要改变受体和配体分子的相对位置,取向以及各自的构象,这类方法计算量较大,一般用于特殊请框架(比如蛋白质结合口袋不足以容纳待对接的小分子)的发分子和小分子的对接,代表性对接程序有Fiber Dock,Flex Dock,Fleksy以及薛定谔软件包中的Induced Fit Docking(IFD)流程等。
分子对接算法(结构匹配搜索):
早期的分子对接模型很简单,假定配体分子都是刚性的,预测能力有限。20世纪90年代后,为了更准确地模拟分子对接问题,人们开发了更复杂的程序。这些程序采用三种不同的策略将柔性的配体放入蛋白质的刚性结合口袋。
1.随机法
随机生成配体分子的构象,并将配体在蛋白质的结合位点内进行随机平移和旋转,然后通过打分函数评价产生的各种姿态。AutoDock使用模拟退火来寻找好的姿态,AutoDock Vina是AutoDock4的增强版本,它提高了配体结合模式预测的准确性,并且降低了运行时间。首先,一组初始姿态被生成、打分和局部优化。类似于蚂蚁沿着信息素的路径找到食物,蚁群算法总是优先选择在之前迭代中评分较高的那些姿态的平移,旋转和扭转,从而对姿态迭代优化。
2.基于片段的方法
将配体在其可旋转键位置切割成小片段,以便更好地处理配体构象的柔性。在放置初始片段后,配体在蛋白质的活性位点被重建,过程中每个片段都会被评分。与模拟退火和遗传算法等随机方法不同,构建过程是确定性的,因此对接结果是可以重现的。在逐步构建配体的过程中,使用贪婪算法,将得分高的解的数量控制在几百个以内。基于已经计算的各个片段姿态的对接打分,可以快速确定最优的片段组合。最后,将柔性连接臂匹配到刚性片段集,形成一个粗糙的配体结合姿态,再通过局部能量最小化对结合姿态进行优化。
3.构象集合法
首先生成配体的多种构象的数据库,其次将这些构象放置到蛋白质的活性位点。采用该策略的对接工具有Glide,FRED,TrixX。Glide通过穷举配体扭角空间中的能量极小值,生成各种构象异构体。蛋白质活性位点对这些低能构象进行快速预筛选。预筛选得到的最优姿态首先经过能量最小化优化,然后采用蒙特卡罗方法发现附近扭角空间的极小值。最后,结合打分函数、分子力学计算和配体应变能计算,确定最佳姿态。对于每个构象及蛋白质活性位点,计算一个基于形状和相互作用的描述符。
分子对接的打分函数:
打分函数是蛋白质-配体非共价相互作用强度预测的数学表达式(打分函数被应用在三种不同的场景:对接过程中的姿态预测,虚拟筛选实验中的化合物排序、结合亲和力的预测)。
姿态预测的挑战性是从对接算法提供的众多候选姿态中确定配体在蛋白活性位点内的天然结合模式;而在虚拟筛选中对化合物进行排序,将活性化合物与非活性化合物分离开,是打分函数的主要应用;而结合亲和力预测则是对打分函数要求最苛刻的应用场景,因为必须考虑许多不同的相互作用、效应及它们之间的平衡。
打分函数目前在姿态预测方面表现不错,但在排序和结合亲和力预测方面仍然需要改进。
打分函数的物理化学基础是预测蛋白质—配体结合过程吉布斯自由能的变化。两个分子结合形成复合物时,复合物的能量低于两个独立分子的能量之和。
![]()
焓变不分主要由配体和蛋白质之间的范德华力,静电和氢键相互作用贡献,是打分函数考虑的主要部分。相比之下,熵变部分难以模拟和计算,常常被忽略。熵变主要包括两个方面:从结合位点释放后变得无序的水分子,结合导致蛋白质与配体分子的扭转角被固定。
通常打分函数分为三种不同的类型:
1.基于力场的打分函数
使用经典的分子力场来估算蛋白质—配体复合物中的相互作用。分子力场(如适用于蛋白质的AMBER和适用于小分子的MMFF)计算的主要能量包括扭转能,伦纳德—琼斯势能和静电能等。基于力场的打分函数缺少解释熵变的项,并且会高估静电相互作用和氢键,因此主要用于姿态预测而非虚拟筛选排序或结合亲和力预测。
2.基于知识的打分函数
受益于数量不断增长的蛋白质—配体复合物共晶结构。根据蛋白质—配体复合物中常见相互作用及距离的统计分析,可以计算出成对原子间的势能,这是此类打分函数的基础。
3.经验打分函数
经验打分函数是最常见的打分函数类型。它用不同的项代表蛋白质—配体复合物中的不同理化效应(如氢键作用,疏水效应或金属相互作用)并求和,计算总的结合自由能。
![]()
经验打分函数的最大缺陷是只有当对接的靶标与校准数据集的蛋白质相近时,表现才更好。
另一种策略是特异地针对一个目标系统来训练打分函数;这被称为定制或靶标特异性的打分函数。
Autodock Vina中是基于经验的打分函数。
利用分子对接方式进行虚拟筛选过程:
在进行筛选之前,先对候选分子进行前过滤(利用一些过滤器对化合物库进行过滤,过滤器会对分子量,logP,氢键供体和受体的数量等分子理化性质进行限制。必要时,还可以采用子结构匹配或基于配体的药效团模型等更复杂的过滤器。这些过滤器可以大大减小需要搜索的化学空间,从而加速虚拟筛选过程)。
第一步,检查待筛选的化合物库中化合物结构是否正确。此外,根据所用对接软件的要求,生成化合物的互变异构体,质子化状态和各种构象。
第二步,指定用于配体对接的结合位点,并优化蛋白质的内部氢键网络,做好对接准备。对接位点中通常需要包括保守水分子和重要辅助因子。
第三步,通过将化合物放置到蛋白质的结合位点来产生姿态。由于每种化合物可能产生的平移,旋转和构象数目是海量的,只可能产生所有可能姿态中的有限的一部分。姿态产生过程可以通过药效团过滤器来指导。
第四步,通过打分函数预测每一个姿态下配体与蛋白质的结合亲和力,进行打分,并根据每个化合物得分最高的姿态对化合物排名。
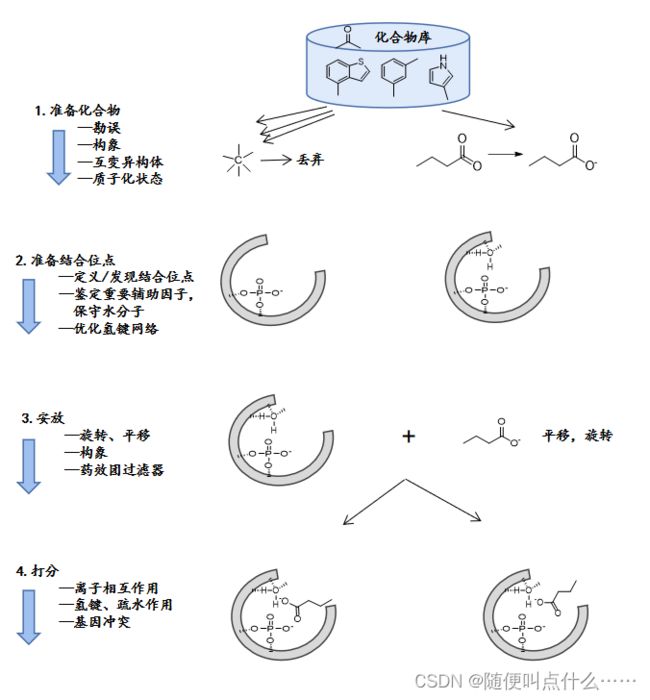
在对接和打分阶段之后也可以使用过滤操作。这些后过滤器通常会限定化合物的潜在结合模式。
最常见的后过滤器是基于蛋白质结构的药效团。
基于蛋白质结构的药效团过滤器描述了化合物能够发挥其活性所需的分子特征。一个特征可以是化合物结合到蛋白质的某个位置所需的某个子结构或理化性质。经过精心设计并通过实验数据验证的药效团过滤器可以在完成对接打分之后进行后过滤。由于每个药效团过滤器可以代表一种不同的结合模式,那些通过同一个药效团过滤器的配体可以被视为具有相似结合模式。(使用药效团过滤器的一个缺点是结果被限制到那些已知结合模式,因此它限制了对接程序发现新结合模式化合物的能力。)