直系同源基因分析(orthofinder方法)
直系同源基因分析(orthofinder方法)
- 直系同源基因判定的方法有两种
- orthomcl这个方法已经是应用很广泛的一个方法了,但是存在问题:
- solve:
- 原理:
- OrthoFinder2 可以推断出正交组,基因树,基因复制事件,有根树种和广泛的比较基因组统计数据,从而实现下一代系统发育学分析。
- 安装orthofinder
- 安装diamond
- 安装mcl
- 安装FastMe
- 测试你是否可以运行OrthoFinder
- Orthofinder --help
直系同源推断在生物科学中具有根本重要性,支持系统发育学,比较基因组学和基因功能预测
直系同源基因判定的方法有两种
一种是将物种的直系同源基因两两比较,不同物种的每一对都是直系同源,但存在基因重复,使得用这种方法找到的同源关系是不具有传递性的(A和B是同源的,B和C是同源的,但是A和C不是同源的)multiparanoid和OMA方法都是这个原理。所以需要每个同源基因都在多个集合中存在才行,进行两两比较。
另一种是识别完整的正交群,一个同源组orthogroup包含所有物种共同祖先基因,同时包含直系/旁系同源基因,orthomcl是这个原理,它是通过正交组做推断,用BLAST计算多个物种序列之间的序列相似性得分,然后用MCL聚类算法,识别该数据集中高度相似的序列组。
orthomcl这个方法已经是应用很广泛的一个方法了,但是存在问题:
- 在BLAST看序列相似度时发现,长序列比短序列更准确,有时会留下较长的质量差的序列,而丢弃较短的单质量好的序列。
- orthoBench是唯一可用的公开的正交群标准数据集,用orthoBench数据集评估orthoMCL性能,发现它对序列长度有依赖性,太长或者太短的序列都不准确。
- recall率有待提高。
solve:
- orthoFinder解决了这个问题,它将BLAST评分做了转换,将原来的e值准换成bit值(比特值)来减少序列长度对准确度的影响,因为e值存在阈值,e 小于10的-180次方的都被算作0,而比特值没有阈值。
- orthoFinder用RBNH代替了传统的RBH提高了recall。
原理:
它的基本原理,首先all vs all blast 选best hits Top5%,用比特值代替e值,用最小二乘法将Top5%的分数值拟合成一个线性模型,将比特值用这个模型转换之后,较长的质量差的hit将不再优于短的质量好的序列,然后将基因长度和系统发育距离标准化,用的是RBNH方法,传统的是用RBH方法,它是一种高精度鉴定直系同源基因对的方法,用BLAST best hit的倒数做的,但是RBNH是用标准话的比特值来做的。
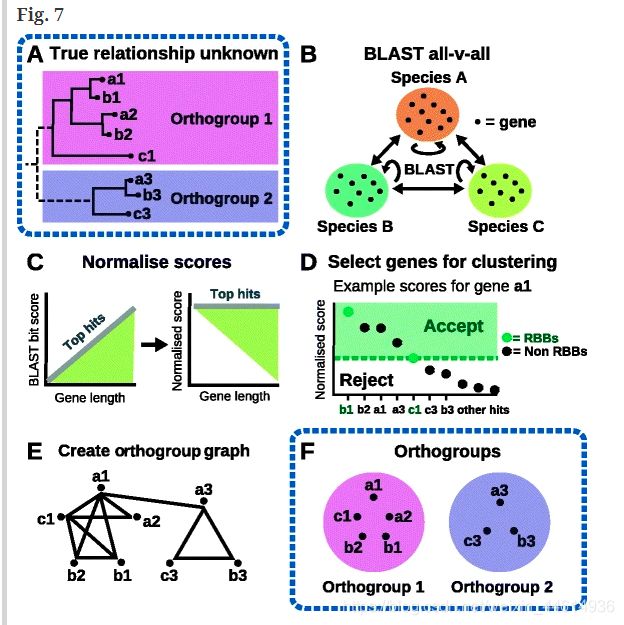
原理:
- BLAST all-vs-all,使用BLASTP以e =10e-3进行搜索,推断同源基因(还可以选择DIAMOND和MMSeq2)。
- 基于基因长度和系统发育距离对BLAST bit得分进行标准化。
- 使用RBNH方法,确定一个同源组序列性相似度的阈值,筛选出质量好的直系同源组(orthogroup graph)
- 构建直系同源正交组,通过归一化的比特值画出正交组的边缘连接,用作MCL的输入
- 使用MCL算法对基因进行聚类,划分直系同源组
再用orthoBench数据集做测试,发现长度依赖性明显减少。而且recall和F值明显高于orthoMCL方法,精准度稍有降低,但整体高,假阳性、假阴性都优于其他方法。而且适用于分析不完整的数据集,且比其他的方法更快速,可以扩展到其他几百个物种中使用。
orthoFinder饰用Python编的,它需要Python,numpy和scipy库,还要把BLAST+和MCL算法安装到orthoFinder。
OrthoFinder2 可以推断出正交组,基因树,基因复制事件,有根树种和广泛的比较基因组统计数据,从而实现下一代系统发育学分析。
主要由三个主要阶段组成:第一阶段,推测正交群; 第二,物种根系和基因树的推断; 第三,来自这些有根基因树的直系同源物和基因复制事件的推断。
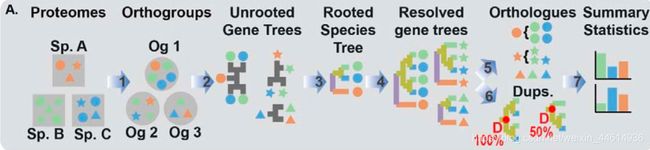
最快的版本使用DIAMOND 进行序列相似性搜索,同时使用DendroBLAST 提供正交群推断和基因树推断的原始数据。使用STAG和STRIDE 10算法实现物种树的推断。为了避免由于谱系排序不完整和基因树错误而导致的低同源序列recall,使用DLCpar强大的复制-丢失-合并模型(duplication-loss-coalescent )的一个增强的、更可伸缩的版本,从根基因树中推断出同源性。
搜索同源序列和建树可以用其他的方法(如BLAST 或MMseqs2 )来替代DIAMOND。OrthopFinder源代码和可执行文件可从 https://github.com/davidemms/OrthoFinder获得。OrthoFinder解决了直系同源分析的准确性和可扩展性等方面的几个关键技术挑战。它在速度和直系同源recall方面取得了实质性进展。
安装orthofinder
https://github.com/davidemms/OrthoFinder/releases/tag/v2.2.7
安装diamond
wget https://github.com/bbuchfink/diamond/releases/download/v0.9.22/diamond-linux64.tar.gz
tar xzf diamond-linux64.tar.gz
mkdir ~/bin
cp diamond ~/bin
export PATH=$PATH:~/bin/
#如果上面的环境不行就这样:
vim ~/.bashrc
echo ‘export PATH=/home/zhuqh/xx/:$PATH’ >>~/.bashrc
source ~/.bashrc
安装mcl
http://micans.org/mcl/
cd mcl
mkdir bin
./configure --prefix=/home/zhuqh/xx/mcl/bin
#编译
make
make install
cd bin
echo ‘export PATH=/home/zhuqh/xx/mcl/bin/bin:$PATH’ >>~/.bashrc
source ~/.bashrc
安装FastMe
http://www.atgc-montpellier.fr/fastme/binaries.php
cd fastme
mkdir bin
./configure --prefix=/home/zhuqh/xx/fastme/bin
#编译
make
make install
cd bin
echo ‘export PATH=/home/zhuqh/xx/fastme/bin/bin:$PATH’ >>~/.bashrc
#(找到一个别的编译方式:
./configure --prefix=~/opt/biosoft/mcl-14.137
make -j 20 && make install )
测试你是否可以运行OrthoFinder
OrthoFinder-2.2.7/orthofinder –h
如果要将orthofinder可执行文件移动到另一个位置,则还必须将附带的config.json文件放在同一目录中。
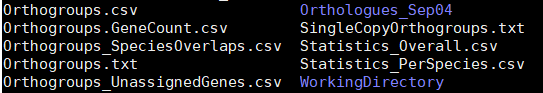
结果文件
运行结束后,会在ExampleData里多出一个文件夹,Results_Feb14, 其中Feb14是我运行的日期
直系同源组相关结果文件,将不同的直系同源基因进行分组
·Orthogroups.csv:用制表符分隔的文件,每一行是直系同源基因组对应的基因。
·Orthogroups.txt: 类似于Orthogroups.csv,只不过是OrhtoMCL的输出格式
·Orthogroups_UnassignedGenes.csv: 格式同Orthogroups.csv,只不过是物种特异性的基因
·Orthogroups.GeneCount.csv:格式同Orthogroups.csv, 只不过不再是基因名信息,而是以基因数。
直系同源相关文件,分析每个直系同源基因组里的直系同源基因之间关系,结果会在Orthologues_Feb14文件夹下,其中Feb14是日期
·Gene_Trees: 每个直系同源基因基因组里的基因树
·Recon_Gene_Trees:使用OrthoFinder duplication-loss coalescent 模型进行发育树推断
·Potential_Rooted_Species_Trees: 可能的有根物种树
·SpeciesTree_rooted.txt: 从所有包含STAG支持的直系同源组推断的STAG物种树
·SpeciesTree_rooted_node_labels.txt: 同上,只不过多了一个标签信息,用于解释基因重复数据。
比较基因组学的相关结果文件:
·Orthogroups_SpeciesOverlaps.csv: 不同物种间的同源基因的交集
·SingleCopyOrthogroups.txt: 单基因拷贝组的编号
·Statistics_Overall.csv:总体统计信息
·Statistics_PerSpecies.csv:分物种统计信息
STAG是一种从所有基因推测物种树的算法,不同于使用单拷贝的直系同源基因进行进化树构建。
如果你想根据多序列联配(MSA)结果按照极大似然法构建系统发育树,那么你需要加上-M msa。这样结果会更加准确,但是代价就是运行时间会更久,这是因为OrthoFinder要做10,000 - 20,000个基因树的推断。
Orthofinder --help
简单的用法: 正交查找器[选项]-f 将中的新物种添加到 正交查找器[选项]-f 选项: -t 并行序列搜索线程数[默认= 16] -M 基因树推断方法。选项“dendroblast”和“msa” (默认= dendroblast) -S 序列搜索程序[默认= blast] 选项:blast, mmseqs, blast_gz, diamond -A 选择:muscle,mafft -T 选项:iqtree、raxml-ng、fasttree、raxml -s <文件>用户指定的根物种树 -I MCL通胀参数[Default = 1.5] -x Info用于以othoXML格式输出结果 -p -l只执行单向序列搜索 -n 名称以附加到结果目录 -h打印帮助文本 工作流停止选项: -op 在为BLAST准备输入文件后停止 -og 在推断出正交群后停止 -os 操作系统在为正交组编写序列文件后停止 (要求- m msa) -oa停止后,推断对齐的正交组 (要求- m msa) -ot 在推断基因树为正群后不要停止 工作流启动命令: -b -fg -ft 许可: 根据GNU通用公共许可证(GPLv3)发布。看到License.md 引用: 当发表使用正交器的作品时,请注明: Emms D.M. & Kelly S. (2015), Genome Biology 16:157
在
-a 并行分析线程数[默认值= 1]