BioRxiv|利用AlphaFold进行环肽结构预测和设计-Baker课题组环肽新工作

个人感觉:作者 Gaurav Bhardwaj是大佬David Baker 课题组专门研究环肽的专家,rosetta环肽的结构预测以及设计模块基本都有他的参与,无论是对称环肽还是透膜性的设计都做的非常好。目测这篇文章应该也是朝着Cell/Nature 这类期刊准备的。从偏计算的Rosetta,转到AI-driven的Alphafold可谓是一个惊喜,不知道环肽的设计是否可以由这一篇进入更广大的AI-driven领域。其中使用的Rosetta能量景观和幻觉环肽设计都是之前的工作,这篇文章很好地集合了之前的工作基础,实属精品。
题目:Cyclic peptide structure prediction and design using AlphaFold
文献来源:https://doi.org/10.1101/2023.02.25.529956
代码:Example scripts for structure prediction, sequence design, and hallucination are available at https://github.com/sokrypton/ColabDesign/blob/main/af/examples/af_cyc_design.ipynb Rosetta software suite can be downloaded from https://www.rosettacommons.org
内容:
1.背景介绍
Alphafold、RoseTTAFold等深度学习模型近几年在蛋白质预测领域大放异彩。但是这些模型往往只适用天然氨基酸组成的较大氨基酸。虽然,其在预测小肽以及肽-蛋白复合物的结构方面的应用也比较有限,但是作者认为应该尝试利用这些算法进行环肽结构的预测。大环化在生物活性天然产物和治疗性肽发现运动中很常见,因为它具有一些结构、稳定性和渗透性优势。肽中缺乏自由末端使它们对外蛋白酶和肽酶具有更强的抵抗力。尽管这类化合物缺乏规则的二级结构,环具有的约束可以将小肽锁定在稳定的折叠结构中。虽然Rosetta已经被长期应用于环肽的设计和结构的预测,但是因为其骨架采样的计算成本较高,因此需要寻找一些计算成本更小的方法进行准确的环肽结构预测。
环肽的高分辨率结构的结构比较少,因此,从头训练一个用于环肽的深度学习模型是非常困难的。作为替代方法,模型可以在Rosetta以及MD(分子动力学方法)等方法的基础上进行训练。然而,这种模型的准确性和性能将受到用于生成训练数据的方法准确性的限制。事实上,像AlphaFold和RoseTTAFold这样的预训练网络可以被修改从而识别环状结构,并进行基准测试以确定它们在预测环蛋白和肽结构方面的准确性。作者之前使用过KIC方法(一种骨架采样方式),它们注意到环肽主要由天然氨基酸motifs和turn类型组成,这在较大蛋白质的环区域也很常见。而Rosetta能量函数来自于大型蛋白质的晶体结构,能够在肽设计过程中正确地捕获这些motifs。此外,最近的基准研究表明,AlphaFold能够预测载脂蛋白中短肽配体的结构和结合态。因此,作者推断,如果能够适当地强制执行环结构约束和对序列所对应的环结构的位置编码不变性,那么在AlphaFold网络中编码的信息将足以准确地预测和设计环结构。在这里,作者描述了一种环化信息编码并且作为AlphaFold的输入位置编码的方法,并测试了这些变化在预测PDB中可用的环肽结构方面的准确性。接下来,作者报告了一种使用AlphaFold重新设计大环主链序列的方法,以提高它们折叠成设计结构的倾向。最后,他们描述了一种从头幻觉设计新环肽的方法,并使用它来列举7-13元环肽丰富多样性。
2.对环肽结构的预测
作者开始通过修改相对位置编码的输入来扩展AlphaFold,使其能够用于环肽的结构预测。对于线性肽,相对位置编码定义了氨基酸之间的序列分离,相邻残基的序列分离为1,N端和c端由肽的长度- 1分隔(图1A)。为了应用环约束,作者应用了一个自定义的N x N cyclic offset矩阵,该矩阵引入了相对位置编码的环化,并将长度为N的肽的末端残基之间的序列分离改变为1(图1B)。这个相对位置编码,经过一次独热编码和线性投影后,被添加到AlphaFold网络的 evoformer module模块中的pairwise feature中。如果没有这种编码,Attention层具有排列和顺序不变性。作者在ColabDesign 框架中实现了这些变化,该框架可以实现结构预测和设计的 AlphaFold。作者将这个新模型命名为AfCycDesign。作者首先用从PDB中随机选择的环肽序列对其进行了测试,发现这些初始测试的结果显示了正确的肽键连接和末端残基的几何形状,但是模型的预测结果没有在肽结构的其余部分引入扭曲。接下来,作者将序列的环表示进行排列作为输入,观察输出预测是否会发生改变。结果显示所有环排列序列的输出结构都非常相似。
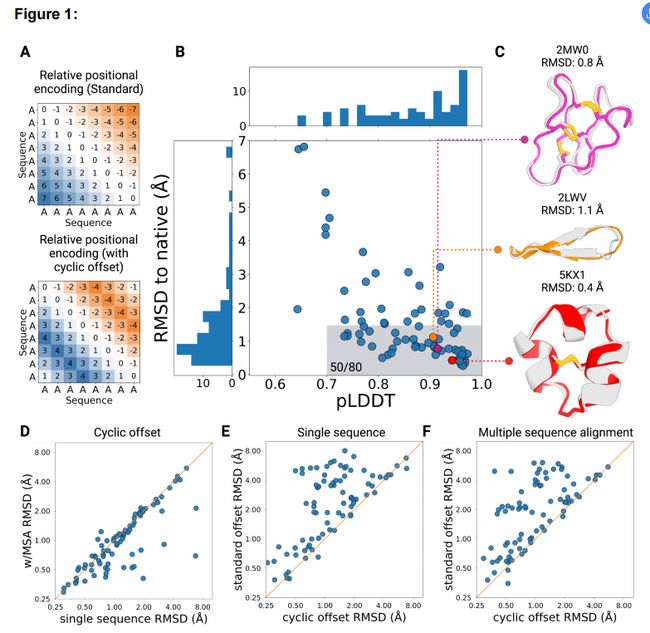
图1 使用AfCycDesign预测天然环肽的结构。(A)一个假设的8元环肽的相对位置编码的例子。AfDesign中的标准编码显示了一个线性肽的残基位置之间的序列分离,其末端是彼此之间的最大距离。在AfCycDesign中应用cyclic offset可以改变这种行为,使肽链两端相互连接。(B)AfCycDesign预测了来自蛋白质数据库的80个环状肽。突出显示的区域涵盖了使用pLDDT > 0.7和RMSD < 1.5埃的良好可信度和准确预测结果。(C)AfCycDysign正确预测的结构结构(RMSD<1.5埃, pLDDT>0.85)。实验确定的结构以灰色表示,而AfCycDesign的预测结构以洋红色、橙色或红色表示。(D)对单个序列的cyclic offset和MSA预测的准确性(RMSDy与实验结构比较获得)的比较。(E)单序列的有无cyclic offset预测的准确性的比较。(F)有无cyclic offset的MSA预测准确性的比较。
接下来,作者评估了AfCycDesign预测PDB中不同环肽结构方面的准确性。他们从PDB中收集了80个典型环肽结构,其序列长度小于40个。这些结构都不在AlphaFold的训练集中,因为该模型训练的时候排除了核磁共振结构和长度小于16个氨基酸的短肽。作者选的这些结构包括了广泛的拓扑结构,具有不同的大小、二级结构、序列和功能。值得注意的是,在测试集中,许多肽,如不同的植物衍生的环肽或环状结蛋白折叠,由多个半胱氨酸残基和二硫键组成。多种二硫键连接的组合-4个半胱氨酸存在3种可能连接,6个半胱氨酸存在15种连接,给之前的计算方法带来了挑战-因为二硫键连接必须明确定义。
AfCycDesign的评价指标:与实验确定结构向比较主链重原子RMSD,以及结构预测置信指标-预测局部距离差异(pLDDT)。总的来说,AfCycDesign的预测与实验确定的结构很接近,中位数pLDDT和RMSD分别为0.88和1.13埃(图1B)。80个测试案例中,有50个预测的结构与实验结构相比,具有良好的可信度(pLDDT > 0.7)而且主链RMSD小于1.5埃。值得注意的是,在49个AfCycDesign判定为高可信度预测结构(plDDT > 0.85)的例子中,73%(n = 36)的主链重原子RMSD< 1.5 埃,这表明pLDDT分数可以作为预测环肽的一种筛选指标。另外,正确预测的结构并不局限于特定的肽或拓扑,可以涉及不同的大小和拓扑如富二硫环肽、小环β折叠s和具有非常短α螺旋motifs的肽(图1C)。在14个例子中,模型预测的结构非常接近实验结构(主链RMSD < 1.5埃);然而,AfCycDesign对这些预测结构打出的可信度较低(pLDDT<0.85)(图1B)。这些结果的较低分数来自于其在NMR数据集中也具有很灵活的loop区域。虽然没有对二硫键没有进行额外的约束,大多数高可信度情况下的键连接都会产生,这对于那些结蛋白、共肽、环肽和其他种类的富含二硫化物的肽来说是一个好消息。在预测过程中添加多序列比对(MSA)可以进一步提高模型预测的准确性,80个结构中有58例被正确预测(主链重原子RMSD < 1.5 埃)可信度良好(pLDDT > 0.7),大部分结构的整体预测准确性有所提高(图1D)。相比之下,单序列或基于MSA的预测中去除cyclic offset,会导致预测正确结构的能力显著降低(图1E-F)。综上所述,AfCycDesign方法在预测具有不同环肽结构方面的良好能力。
3.环状多肽的序列重设计
接下来,作者利用环相对位置编码,用AfCycDesign设计环肽的氨基酸序列。他们推断,这种方法将有助于识别新的氨基酸序列,以改善来自天然多肽或使用其他主链采样方法生成的给定主链折叠倾向。为了实现这一点,作者在之前实现的ColabDesign方法中引入了cyclic offsets。这种方法的目标是找到由AlphaFold预测的可以折叠成所需主链的序列。该方法首先使用AlphaFold网络从一个随机序列中预测分布图,并在随后的每个步骤中迭代优化序列,以最小化在该步骤中预测的结构与期望的主链之间的差异。序列优化是由预测的距离图(包含每对残基的距离分布的张量)和从期望结构中提取的一个结构之间的差异(或分类交叉熵)进行指导的。这被证明是一个很好的方法,可以最大化AlphaFold的置信度和最小化预测结构和期望结构之间的差异。
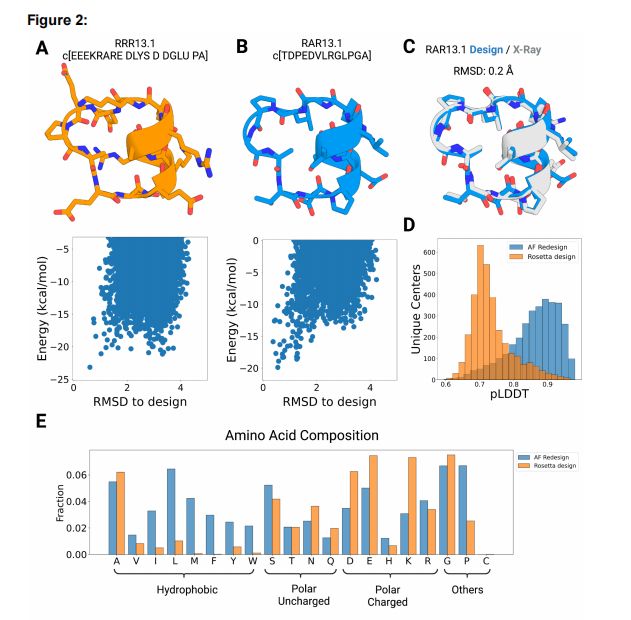
图2 使用AfCycDesign (A)序列设计环肽骨架序列,设计Rosetta设计的13残基环肽RRR13.1。序列中的l-氨基酸用它们的单个字母的编码表示,而d-氨基酸用四个字母的编码表示。用Rosetta环肽预测方法计算的预测能量景观如下所示。由AfCycDesign设计的13-残基环肽RAR13.1的(B)序列和设计模型。序列中的l-氨基酸用它们的单字母编码表示。由Rosetta计算出的预测能量景观如下图所示。(C)对RAR13.1设计构象(蓝色)和高分辨率x射线晶体结构(灰色)的对齐显示有非常接近的匹配,具有0.2埃的Cα RMSD。(D)Rosetta(橙色)和AfCycDesign(蓝色)设计序列的pLDDT评分分布。每个群体都由相同的主链设计,代表3274个独特的13元环肽的结构簇。由Rosetta(橙色)和AfcycDesign(蓝色)设计的序列的每个氨基酸频率,这些序列来自3274个独特的13环肽结构簇的主链。
作者开始设计适合于靶向蛋白质-蛋白质界面上常见的螺旋-螺旋相互作用的肽架构。他们使用Rosetta环肽设计方法生成了13环肽的457,615个骨架,这些骨架都包含一个短的7氨基酸螺旋结构。为了在这次大规模运行中识别所有独特的形状,我们使用扭转箱方法对得到的主干进行聚类:生成一个表示结构的箱子串,每个氨基酸根据ɸ、ψ和ω扭转角分配到一个箱子;箱A和B指拉马钱德兰图的α和β区域,而箱X和Y指图的 ɸ, ψ区域的镜像区域。这也是Rosetta设计中常用的一种方式。箱字符串的环排列也被分组到相同的结构簇中。作者获得了29,249个具有独特的箱子串簇,并选择了一个主链RRR13.1(箱序列:AAAAAAXBYBBAB)进行重新设计,因为Rosetta设计的同一主链序列在其能量景观中有一个小的能量间隙(ΔE<2千卡/摩尔)(图2A)。事实上,AfCycDesign设计的序列与Rosetta设计的序列有显著差异,序列中有12个突变,肽核心只有一个丙氨酸被保留。作者首先评估了AlphaFold设计的序列RAR13.1的性质。他们通过使用Rosetta的环肽预测方式计算了其能量景观。AlphaFold序列作为其最低能量构象收敛于设计结构,在设计结构和备选构象之间的能量间隙更大(ΔE ~ 6.0千卡/mol)(图2A ,B)。为了验证Alphafold设计的序列是否能够折叠成所需要的结构,外消旋高分辨率x射线晶体学确定了其三维结构,并将其与计算设计的构象进行了比较。x射线晶体结构与设计模型非常接近,Cα RMSD为0.2埃,x射线晶体结构中13个侧链转子中有10个与设计模型所给出的相匹配(图2C)。
鉴于Alphafold设计的RAR13.1的成功结构验证,作者决定重新设计3274个独特结构簇中的代表性肽,这些结构从大规模主干取样运行中选择,含有聚丙氨酸(或d-丙氨酸)的主链Rosetta能量小于0千卡/摩尔。同时,作者也用Rosetta设计了选定的主链,并将它们与AfCycDesign生成的序列进行了比较。正如预期的那样,同样的主链,AfCycDesign设计的序列比Rosetta设计的序列具有更好的pLDDT评分分布(图2D)。而在Rosetta设计的序列中,只有63个类的pLDDT>0.9;AfCycDesign序列中有1145个类的pLDDT>0.9(图2D)。然而,作者不得不将Rosetta的设计方法限制在典型的20个氨基酸上(如pLDDT计算所需要的)-以前他们可以进行异手性设计。除了比较结构预测置信度指标外,作者还研究了由AfCycDesign和Rosetta设计的序列的氨基酸组成和化学性质的差异。与Rosetta设计的相同主链序列相比,AfCycDesign设计的序列通常更疏水,包含更多的脯氨酸(图2E)。总的来说,我实验结果表明,AfCycDesign可以用来设计可折叠成所需结构的环肽骨架序列。更广泛地说,AfCycDesign方法是对其他肽主链生成方法的补充,可以与这些方法结合,快速找到预测在一系列拓扑中正确折叠的序列。
4.幻觉环状肽的从头设计
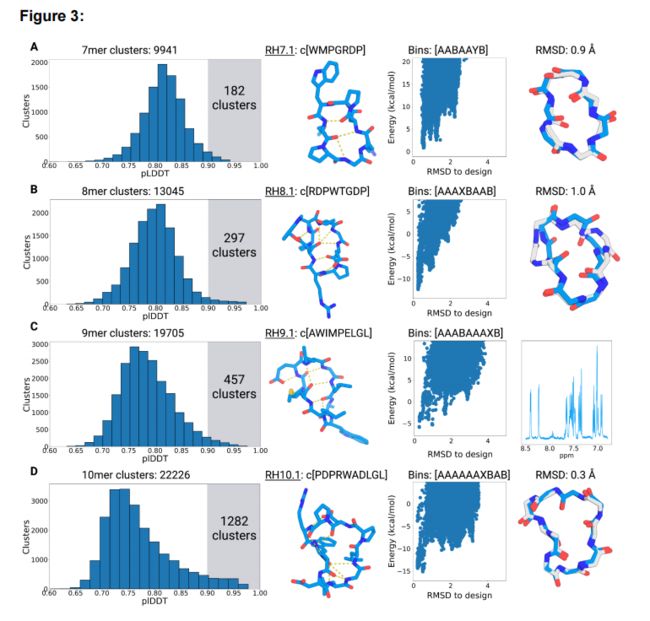
图3 使用AfCycDesign设计了7-10元幻觉环肽。结构预测置信度指标(pLDDT)的分布和从(A) 7元环、(B) 8元环、(C) 9元环和(D) 10元环的大规模采样中选择候选物的验证。对于每一行,第一列描述了从48000个7-10残基的所有独特结构簇的pLDDT分数的分布。每个大小的集群数量在图上描述。每个图中突出显示的区域显示了pLDDT评分>为0.9的集群数量。第二列显示了用于结构表征的模型的幻觉结构和序列。氢键用黄色虚线表示。第三列显示了Rosetta计算出的选定的幻觉模型的能量景观。每个散点(蓝色)表示相同设计序列的不同构象。所选设计模型的扭转箱串显示在图的顶部。第四列显示了在幻觉构象(蓝色)和x射线晶体结构(灰色)之间的align。RH9.1的一维核磁共振谱显示。
接下来,作者开发了一种环肽的幻觉方法(hallucination),同时对序列和结构进行采样,并将其应用于枚举不是之前重设计方法生成的13肽螺旋主链之外的大环肽。他们实现了一个三步幻觉管道,用于生成序列,预测其折叠成高度可信的结构环肽。该方法以损失为指导,试图提高预测置信度指标pLDDT和预测对齐误差(PAE),以及分子内接触的数量。
作者从由7-10元环肽开始,并为每种大小的环列举了48,000个幻觉构象。然后使用前面描述的基于扭转箱的聚类对这些大采样运行的结构进行聚类,并鉴定出9941个7元环肽、13405个8元环肽、19705个9元环肽和22206个10元环肽(图 3A)。在所有独特的结构簇中,7、8、9和10元环肽分别有182、297、457和1282个簇,至少有一个结构被预测可折叠成设计结构(pLDDT>0.9)(图3A)。鉴于在天然结构预测和重新设计方面的结果,作者期望通过0.9严格置信度量的肽能够正确地折叠到设计的结构中。因此他们选择这些序列进一步通过Rosetta环肽结构预测方法进行验证。为了评估这些序列的折叠倾向,计算Pnear值(描述能量景观的一种指标)。Pnear值范围为0到1,值为1表示所设计的结构是该序列的单一最低能量构象。在这些计算中,许多幻觉序列显示出良好的折叠倾向,114 个7元环肽、186个8元环肽,139个9元环肽和76个10元环肽显示Pnear值大于0.6。作者在每个环肽大小都选择了一个幻觉设计构象,这些设计pLDDT > 0.9和Pnear > 0.9。作者使用这些设计构象进行进一步的实验验证和结构表征。所有四种选定的设计构象都缺乏规则的二级结构,但通过广泛的分子内主链到主链和主链到侧链的氢键来稳定。RH7.1、RH8.1、RH9.1和RH10.1的构象分别为3、5、5和6个分子内氢键(图3,第二列)。所选模型的整体形状也由典型的α、β和γ转弯的组合来指导。构象RH7.1由一个I型β转弯和一个重叠的γ和α转弯组成,所有的转弯都由脯氨酸残基成核。构象RH8.1包括两个I型β转弯,由i + 2的i位天冬氨酸残基的侧链到主链氢键稳定。构象RH9.1还包含两个I型β转弯,由蛋氨酸-4和亮氨酸-9之间的一对长程氢键隔开。构象RH9.1的疏水性很显著,在设计中唯一的极性残基是单一的谷氨酸。RH10.1的序列也具有显著的疏水性,有多个暴露的非极性侧链,以及色氨酸和一个亮氨酸之间的疏水性堆积,这稳定了一个分子内氢键相互作用很少的区域。作者还在选定的设计构象中观察到多个甘氨酸和脯氨酸,脯氨酸提供了构象约束,甘氨酸进入拉马钱德兰图的X和Y箱(phi角>0度)。
作者试图结晶所有四种肽,并获得了7元环肽、8元环肽和10元环肽的高分辨率x射线晶体结构(图3,第四列)。RH7.1的x射线结构与幻觉构象非常接近,两个结构之间的Cα RMSD为0.9 埃。设计构象和β射线晶体结构之间有很小的差异,β射线晶体结构具有一个来自天冬氨酸侧链的额外氢键,稳定了没有设计的I型转弯。相比之下,RH8.1结构更偏离幻觉构象,Cα RMSD为1.0 埃,脯氨酸-3和甘氨酸-6的扭转在设计的构象与x射线晶体结构不同。这种差异在序列中的单一甘氨酸位置最为显著,在那里ɸ扭转被翻转。RH10.1的结构与幻觉构象基本一样,Cα RMSD为0.3埃。晶体结构中的侧链转子也与设计模型非常匹配,两个亮氨酸和天冬氨酸是相同的。
作者无法得到RH9.1的x射线结构;然而,该设计的一维核磁共振显示出尖锐和分散的峰,表明它也是折叠的(图3C,第四列)。需要进一步的结构表征来确定折叠态是否与设计模型相匹配。
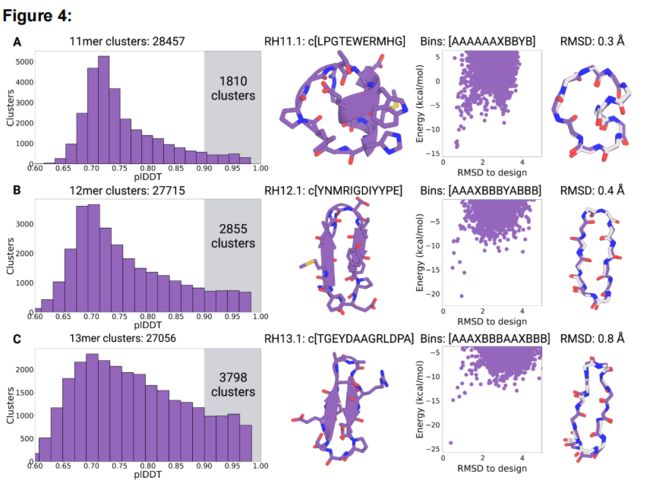
图4:使用AfCycDesign设计11-13元幻觉环肽。结构预测置信度指标(pLDDTs)的分布和从(A) 11(B) 12和(C) 13环肽的大规模抽样中选择的候选物验证。对于每一行,第一列描述了从48000个11-13元幻觉环肽中识别出的所有独特结构簇的pLDDT分数的分布。每个大小的唯一集群的总数在图标题描述。每个图中突出显示的区域显示了pLDDT评分>0.9的集群数量。第二列显示了用于结构表征的模型的幻觉结构和序列。第三列显示了Rosetta计算出的选定的幻觉构象的能量景观。每个散点表示相同设计序列的不同构象。所选设计模型的扭转箱串显示在图的顶部。第四列显示了幻觉模型(紫色)和x射线晶体结构(灰色)之间的align。
接下来,作者进行11-13个氨基酸组成的大环肽的幻觉设计。在基于Rosetta的方法的尝试中,设计大型结构化环肽是一个非常重大挑战,该任务需要额外的二硫键来稳定它们。作者想知道AfCycDesign是否可以在不需要额外键的情况下产生这个大小范围内的大环肽。针对11、12和13环肽,作者从大规模设计计算中分别鉴定出28457、27715和27056个独特的结构簇(图4,第一列)。实验获得了相当数量的簇,且这些簇的结构被预测可以折叠成设计的结构:11、12和13环肽分别有1810、2855和3798个簇,这些簇构象的AlphaFold pLDDT > 0.9。作者选择了一个pLDDT > 0.9和Pnear > 0.9的序列进行实验验证。与较小的7-10个氨基酸设计构象不同,作者从11-13个氨基酸中选择了3种设计中典型二级结构的短motifs(图4,第二列)。设计的RH11.1包含一个8个残基的α-螺旋motifs,RH12.1和RH12.1具有较短的扩展β片。值得注意的是,序列长度为12和13的环肽-RH12.1和 RH13.1都具有非典型大小的β环链,这种结构在6、10和14环肽上很受青睐。这些环肽结构还包括广泛的分子内氢键,在11、12和13环肽设计构象中分别有9、7和9个氢键。RH11.1中的短螺旋motif通过一个具有四个氨基酸的扩展loop进行环化,并具有一个由苏氨酸残基介导的n端螺旋盖帽motif(图4A,第二列)。RH12.1是一个短的β片,典型的类型II‘ β-turn在一端连接链,另一端连接α转。在RH13.1,链配对进行移位因为氢键从天冬氨酸侧链骨干酰胺氮,创建一个扭曲β-折叠环化两端类型β和α,并通过非极性侧链之间的疏水相互作用进一步稳定(图4,第二列)。
我们利用固相化学肽合成技术合成了RH11.1、RH12.1、RH13.1及其镜像,并利用外消旋x射线晶体学测定了这三种肽的结构。这三种多肽段的高分辨率晶体结构与它们的幻觉构象非常匹配,RH11.1、RH12.1和RH13.1的Cα RMSDs分别为0.3、0.4和0.8(图4,第四列)。x射线晶体结构中的转向类型和氢键模式也与所有三种肽的设计构象非常匹配。在RH13.1中观察到的较大的RMSD是由于沿着肽链传播的较小的扭转偏差,而不是之前看到的RH7.1和RH8.1的反过来类型或翻转酰胺的变化。设计中大部分的关键侧链相互作用也在x射线结构中观察到;两个最明显的偏差一是RH11.1的色氨酸翻转180°,但环还是和设计构想一样与组氨酸堆积,二是RH12.1的酪氨酸与精氨酸形成cation-π作用而非与主链产生作用。综上所述,这些数据说明了AfCycDesign在环肽的从头幻觉的高度准确度。该方法可以进行11-13元大环的设计,并且不需要额外的二硫键来稳定先前提出的结构。更广泛地说,这里描述的幻觉方法和广泛的结构抽样提供了一些新的骨架。
5 讨论
作者报告一种方法将环化相对位置编码AlphaFold网络和利用它开发几个关键应用的计算方法,包括结构预测环肽序列,重新设计氨基酸自然和设计环肽骨架,和不同的序列,大小和拓扑的环肽幻觉设计。作者对PDB中的环肽进行结构预测的测试,体现了具有offset的AlphaFold的显著准确性;与实验结构相比,80个序列中有50个的RMSD< 1.5 埃和pLDDT > 0.7。在49个高置信度(pLDDT > 0.85)的例子中,73%基本可以与实验结构相似(RMSD < 1.5)。因此作者这种方式有留意观察自然环肽结构,并使更好的过滤设计的肽从而获得正确折叠的结构。
作者还描述了AfCycDesign,一种重新设计环肽主链序列的计算方法,AfCycDesign设计的序列比Rosetta方法生成的序列具有更好的plddt和折叠倾向。作者比较AfCycDesign序列和Rosetta设计的序列,发现了一些明显差异,包括AfCycDesign增加了疏水和构象限制性的氨基酸的使用。作者进一步AfCycDesign同时进行幻觉序列和结构设计,枚举了数千上万的独特的7-13元环肽结构集群肽。其中包括了10681独特的簇,这些簇被模型认为非常有可能折叠成设计结构(pLDDT > 0.9)。重新设计的幻觉环肽的x射线晶体结构显示了方法的可靠性:所有7个x射线晶体结构(一个重新设计和六个幻觉设计)非常接近他们的设计构象-RMSDs不到1.0。值得注意的是,幻觉方法可以设计11-13的大环肽,这个任务如果没有额外的交联键添加是非常难的。此外,作者之前注意到L-和d-氨基酸模式在生成结构环肽中的重要性;然而,这里描述的幻觉肽违反了这些指南-环肽:只有L-氨基酸,但折叠良好。作者承认d-氨基酸和其他非典型氨基酸所提供的蛋白酶和代谢稳定性的好处,并相信这项工作为未来开发深度学习网络提供了基础,该网络可以在设计过程中纳入更广泛的化学多样性。
幻觉肽预计可以折叠成其设计的结构(pLDDTs > 0.9),以及它们的镜像,具有丰富的功能如目标结合和跨膜。结合相互作用可以通过蛋白质-蛋白质相互作用的嫁接motifs或从头设计来整合。目前和未来的努力的一个焦点是扩展计算方法的幻觉环肽结合物针对治疗目标。在过去的五年里,深度学习方法已经导致了治疗性蛋白质设计的巨大进步。通过本文提出的计算方法,类似的进展可以扩展到具有高治疗意义的结构环肽的定制设计。
-------------------------------------------
欢迎点赞收藏转发!
下次见!