使用kegg数据库的一点见解
使用kegg数据库的一点见解
对kegg这个数据库感兴趣主要是看到Jimmy之前在推文中讲到做GSEA分析还是用kegg原始数据库好,因为msigdb这个数据库关于通路的数据是比较老的。而另外一个原因就是这个数据库确实很重要,而之前也没去好好研究过,就一两句代码就算完事了。
首先我看到y叔的clusterprofiler这个包的源代码,因为这个包是普遍接受用来做富集分析的。
enrichKEGG <- function(gene,
organism = "hsa",
keyType = "kegg",
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
universe,
minGSSize = 10,
maxGSSize = 500,
qvalueCutoff = 0.2,
use_internal_data = FALSE) {
if (inherits(organism, "character")) {
species <- organismMapper(organism)
if (use_internal_data) {
KEGG_DATA <- get_data_from_KEGG_db(species)
} else {
KEGG_DATA <- prepare_KEGG(species, "KEGG", keyType)
}
} else if (inherits(organism, "GSON")) {
KEGG_DATA <- organism
species <- KEGG_DATA@species
} else {
stop("organism should be a species name or a GSON object")
}
res <- enricher_internal(gene,
pvalueCutoff = pvalueCutoff,
pAdjustMethod = pAdjustMethod,
universe = universe,
minGSSize = minGSSize,
maxGSSize = maxGSSize,
qvalueCutoff = qvalueCutoff,
USER_DATA = KEGG_DATA)
if (is.null(res))
return(res)
res@ontology <- "KEGG"
res@organism <- species
res@keytype <- keyType
return(res)
}
get_KEGG_Env <- function() {
if (! exists(".KEGG_clusterProfiler_Env", envir = .GlobalEnv)) {
pos <- 1
envir <- as.environment(pos)
assign(".KEGG_clusterProfiler_Env", new.env(), envir=envir)
}
get(".KEGG_clusterProfiler_Env", envir = .GlobalEnv)
}
通过观察KEGG的数据是通过下面这两句得到的
if (use_internal_data) {
KEGG_DATA <- get_data_from_KEGG_db(species)
} else {
KEGG_DATA <- prepare_KEGG(species, "KEGG", keyType)
}
- 先来看第一个代码。里面有一个函数
get_data_from_KEGG_db.
开始研究
然后我就发现生信媛19年就开始研究过这个问题,果然每个问题互联网都能给你答案。
KEGG数据本地化,再也不用担心网络问题了 (qq.com)
get_data_from_KEGG_db <- function(species) {
PATHID2EXTID <- as.list(get_KEGG_db("KEGGPATHID2EXTID"))
if (!any(grepl(species, names(PATHID2EXTID)))) {
stop("input species is not supported by KEGG.db...")
}
idx <- grep(species, names(PATHID2EXTID))
PATHID2EXTID <- PATHID2EXTID[idx]
PATHID2EXTID.df <- stack(PATHID2EXTID)
PATHID2EXTID.df <- PATHID2EXTID.df[, c(2,1)]
PATHID2NAME <- as.list(get_KEGG_db("KEGGPATHID2NAME"))
PATHID2NAME.df <- data.frame(path=paste0(species, names(PATHID2NAME)),
name=unlist(PATHID2NAME))
build_Anno(PATHID2EXTID.df, PATHID2NAME.df)
}
get_KEGG_db <- function(kw) {
annoDb <- "KEGG.db"
suppressMessages(requireNamespace(annoDb))
eval(parse(text=paste0(annoDb, "::", kw)))
}
get_data_from_KEGG_db()使用get_KEGG_db()函数从"KEGG.db"中获取了"KEGGPATHID2EXTID"和"KEGGPATHID2NAME"这两个AnnDbBimap。
执行的是get_KEGG_db("KEGGPATHID2EXTID") get_KEGG_db("KEGGPATHID2NAME")这两条命令
具体来说,对于get_KEGG_db(“KEGGPATHID2EXTID”)其实就是执行KEGG.db::KEGGPATHID2EXTID这一条命令。as.list(get_KEGG_db(“KEGGPATHID2EXTID”))就是将KEGG.db::KEGGPATHID2EXTID这一条命令返回的结果转成list。
上面这些随便了解一下就好了,具体要怎么使用可以看下下面这篇文章。
构建自己的R包–KEGG.db - 简书 (jianshu.com)
#安装Y叔的包,
#安装创建KEGG数据库的包的包
remotes::install_github("YuLab-SMU/createKEGGdb")
#创建自己的物种的包create_kegg_db,会自动创建名称为KEGG.db_1.0.tar,gz的包。物种名称的简写,在
b=createKEGGdb::create_kegg_db('hsa')
#安装这个包(默认的包的路径在当前工作目录,根据实际情况修改路径)
install.packages("./KEGG.db_1.0.tar.gz",repos=NULL,type="source")
最终得到KEGG.db_1.0.tar.gz解压后

在inst里有一个sqlite格式的文件(这个格式的文件的作用可以自行百度)。可以看到里面有我们要的两个文档。
library(KEGG.db)
ego <- enrichKEGG(
gene =df$ENTREZID,#enrich这个函数决定只能使用entrezid进行分析,转化这一步很多教程就不写了
keyType = "kegg",
organism = 'hsa',
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
qvalueCutoff = 0.05,
use_internal_data = TRUE#这个参数默认是FALSE,即使用联网数据
)
设置use_internal_data = TRUE,enrichKEGG这个函数就会自动使用KEGG.db的数据,离线也可以使用。最终得到下面的结果,有23条通路(正常不会这么少的,因为我只是为了验证,所以拿了个数据集)
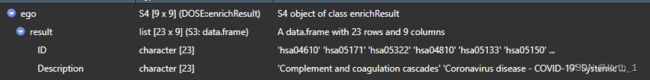
- 第二个代码
prepare_KEGG也看一下源代码
prepare_KEGG <- function(species, KEGG_Type="KEGG", keyType="kegg") {
kegg <- download_KEGG(species, KEGG_Type, keyType)
build_Anno(kegg$KEGGPATHID2EXTID,
kegg$KEGGPATHID2NAME)
}
download_KEGG <- function(species, keggType="KEGG", keyType="kegg") {
KEGG_Env <- get_KEGG_Env()
use_cached <- FALSE
if (exists("organism", envir = KEGG_Env, inherits = FALSE) &&
exists("_type_", envir = KEGG_Env, inherits = FALSE) ) {
org <- get("organism", envir=KEGG_Env)
type <- get("_type_", envir=KEGG_Env)
if (org == species && type == keggType &&
exists("KEGGPATHID2NAME", envir=KEGG_Env, inherits = FALSE) &&
exists("KEGGPATHID2EXTID", envir=KEGG_Env, inherits = FALSE)) {
use_cached <- TRUE
}
}
if (use_cached) {
KEGGPATHID2EXTID <- get("KEGGPATHID2EXTID", envir=KEGG_Env)
KEGGPATHID2NAME <- get("KEGGPATHID2NAME", envir=KEGG_Env)
} else {
if (keggType == "KEGG") {
kres <- download.KEGG.Path(species)
} else {
kres <- download.KEGG.Module(species)
}
KEGGPATHID2EXTID <- kres$KEGGPATHID2EXTID
KEGGPATHID2NAME <- kres$KEGGPATHID2NAME
assign("organism", species, envir=KEGG_Env)
assign("_type_", keggType, envir=KEGG_Env)
assign("KEGGPATHID2NAME", KEGGPATHID2NAME, envir=KEGG_Env)
assign("KEGGPATHID2EXTID", KEGGPATHID2EXTID, envir=KEGG_Env)
}
if (keyType != "kegg") {
need_idconv <- FALSE
idconv <- NULL
if (use_cached &&
exists("key", envir=KEGG_Env, inherits = FALSE) &&
exists("idconv", envir=KEGG_Env, inherits = FALSE)) {
key <- get("key", envir=KEGG_Env)
if (key == keyType) {
idconv <- get("idconv", envir=KEGG_Env)
} else {
need_idconv <- TRUE
}
} else {
neec_idconv <- TRUE
}
if (need_idconv || is.null(idconv)) {
idconv <- KEGG_convert("kegg", keyType, species)
assign("key", keyType, envir=KEGG_Env)
assign("idconv", idconv, envir=KEGG_Env)
}
colnames(KEGGPATHID2EXTID) <- c("from", "kegg")
KEGGPATHID2EXTID <- merge(KEGGPATHID2EXTID, idconv, by.x='kegg', by.y='from')
KEGGPATHID2EXTID <- unique(KEGGPATHID2EXTID[, -1])
}
return(list(KEGGPATHID2EXTID = KEGGPATHID2EXTID,
KEGGPATHID2NAME = KEGGPATHID2NAME))
}
"KEGGPATHID2EXTID"和"KEGGPATHID2NAME"这两个文件是通过下面这个命令得到的,那又得去研究download.KEGG.Path download.KEGG.Module这两个函数了
if (keggType == "KEGG") {
kres <- download.KEGG.Path(species)
} else {
kres <- download.KEGG.Module(species)
}
KEGGPATHID2EXTID <- kres$KEGGPATHID2EXTID
KEGGPATHID2NAME <- kres$KEGGPATHID2NAME
download.KEGG.Path download.KEGG.Module这两个函数的代码放下面,有兴趣可以去看,最重要的可以看到这个函数的运行是联网的,所以网速不行就别用了。可以先使用createKEGGdb生成本地KEGG.db包。
download.KEGG.Path <- function(species) {
keggpathid2extid.df <- kegg_link(species, "pathway")
if (is.null(keggpathid2extid.df)) {
message <- paste("Failed to download KEGG data.",
"Wrong 'species' or the network is unreachable.",
"The 'species' should be one of organisms listed in",
"'https://www.genome.jp/kegg/catalog/org_list.html'")
stop(message)
}
keggpathid2extid.df[,1] %<>% gsub("[^:]+:", "", .)
keggpathid2extid.df[,2] %<>% gsub("[^:]+:", "", .)
keggpathid2name.df <- kegg_list("pathway")
keggpathid2name.df[,1] %<>% gsub("path:map", species, .)
## if 'species="ko"', ko and map path are duplicated, only keep ko path.
##
## https://www.kegg.jp/dbget-bin/www_bget?ko+ko00010
## https://www.kegg.jp/dbget-bin/www_bget?ko+map0001
##
keggpathid2extid.df <- keggpathid2extid.df[keggpathid2extid.df[,1] %in% keggpathid2name.df[,1],]
return(list(KEGGPATHID2EXTID=keggpathid2extid.df,
KEGGPATHID2NAME=keggpathid2name.df))
}
download.KEGG.Module <- function(species) {
keggmodule2extid.df <- kegg_link(species, "module")
if (is.null(keggmodule2extid.df)) {
message <- paste("Failed to download KEGG data.",
"Wrong 'species' or the network is unreachable.",
"The 'species' should be one of organisms listed in",
"'https://www.genome.jp/kegg/catalog/org_list.html'")
stop(message)
}
keggmodule2extid.df[,1] %<>% gsub("[^:]+:", "", .) %>% gsub(species, "", .) %>% gsub("^_", "", .)
keggmodule2extid.df[,2] %<>% gsub("[^:]+:", "", .)
keggmodule2name.df <- kegg_list("module")
keggmodule2name.df[,1] %<>% gsub("md:", "", .)
return(list(KEGGPATHID2EXTID=keggmodule2extid.df,
KEGGPATHID2NAME =keggmodule2name.df))
}
运用prepare_KEGG将前面同一批数据做一下
ego <- enrichKEGG(
gene =df$ENTREZID,
keyType = "kegg",
organism = 'hsa',
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
qvalueCutoff = 0.05,
use_internal_data = FALSE
)
Reading KEGG annotation online: "https://rest.kegg.jp/link/hsa/pathway"...
Reading KEGG annotation online: "https://rest.kegg.jp/list/pathway"...
Warning messages:
1: In utils::download.file(url, quiet = TRUE, method = method, ...) :
the 'wininet' method is deprecated for http:// and https:// URLs
2: In utils::download.file(url, quiet = TRUE, method = method, ...) :
the 'wininet' method is deprecated for http:// and https:// URLs

将前后两次的结果比较一下,发现完全一样,因此两种方法都可以。
ego_f@result %in% ego_t@result
> ego_f@result %in% ego_t@result
[1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE`
下面看一下Y叔的这段话
在线抓最新数据固然好,但不好重复啊,你分析了之后,过了几个月了,准备投稿,发现之前选定的通路重复不出来了,欲哭无泪怎么办?现在自己打KEGG.db包,也就是你把时间点给固定了,什么时候重新跑,只要你用的还是原来的KEGG.db,你必须就可以重复出来。也就是它给了你可以回到过去的「月光宝盒」。
那要用本地数据还是联网见仁见智了。
clusterProfiler/enrichKEGG.R at master · YuLab-SMU/clusterProfiler (github.com)
构建自己的R包–KEGG.db - 简书 (jianshu.com)
KEGG数据本地化,再也不用担心网络问题了 (qq.com)
clusterProfiler优雅的处理KEGG数据 - 简书 (jianshu.com)
clusterProfiler - emanlee - 博客园 (cnblogs.com)