密度泛函理论-从波函数到电荷密度的求解
1
在许多物理学和工程领域中,取得科学和技术进步关键在于能够从原子或者分子尺寸,理解并调控物质的性质。对于描述原子和分子量子行为的基本关系式(薛定谔方程),密度泛函理论(DFT)是一个非常好的求解方法,而且这些原子和分子结构具有一定实际含义。
**引文检索关键词**
Density Functional Theory
2薛定谔方程
20实际最深远的一个科学进展就是量子力学,该理论经过无数次的实验观测验证了其正确性。
简介明了为方便应用的密度泛函理论推导:
将要描述规则排列的原子集合(可以试着比较一个隔离态的孤立分子,以及一个矿物晶体中的有序原子)性质时候,希望知道的基本信息有这些原子的能量,以及移动这些原子时候,他的能量将如何变化。为了定义一个原子的位置,不仅要知道原子核在哪,还需要知道原子的所有电子在哪。原子核的质子或者中子比电子质量大1800左右。说明电子对环境变化感应更快。
所以把要研究的物理问题分成两部分,第一部分,固定原子核位置,求解描述电子运动的方程组。对于在给定的原子核势场中运动的一系列电子,可以得到能量最低的电子构型,最低能量称为电子基态。
将原子核和电子分为各自数学问题的是born-oppen-heimer近似。如果有M个R1,......RM的原子核,那么可以把基态能量E表示为这些原子核位置的函数,即![]() ,此方程也称为这些原子的绝热势能面。一旦能够计算得到该势能面,就能能够处理上面的问题。
,此方程也称为这些原子的绝热势能面。一旦能够计算得到该势能面,就能能够处理上面的问题。
Schrodinger方程的一个简单形式(与时间无关的非相对论Schrodinger方程),即![]()
方程中H是哈米尔顿算符, 是哈米尔顿量的一套求解,或者说本征值。这些解的每一个
是哈米尔顿量的一套求解,或者说本征值。这些解的每一个![]() ,对应一个相应的本征值
,对应一个相应的本征值 ,这是满足本征方程真实值。哈米尔顿的详细量定义取决于所描述的物理体系。比如盒子中粒子、谐振子等,其哈米尔顿量具有简单形式,可以被精确求解。多电子和多原子的交互作用体系是更为复杂的情形。这种情况下,Schrodinger方程可以写成一种更为完整的形式,即:
,这是满足本征方程真实值。哈米尔顿的详细量定义取决于所描述的物理体系。比如盒子中粒子、谐振子等,其哈米尔顿量具有简单形式,可以被精确求解。多电子和多原子的交互作用体系是更为复杂的情形。这种情况下,Schrodinger方程可以写成一种更为完整的形式,即:
![\left [ -\frac{h^{2}}{2m} \sum_{i=1}^{N}\bigtriangledown _{i}^{2}+\sum_{i=1}^{N}V\left ( r_{i} \right )+\sum_{i=1}^{N}\sum_{}^{j<i}U\left ( r_{i,r_{j}} \right )\right ]\psi =E\psi](http://img.e-com-net.com/image/info8/19afb4f6a2f842a9bba3cd9fcbbf46d2.gif)
m为电子质量。前三项依次为每个电子动能,每个电子和所有原子核之间的作用能,不同电子之间的作用能。对于所选定的哈米尔顿量,是电子波函数,是N个电子每个电子空间坐标的函数,即
![]() ,E是电子的基态能量。基态能量和时间无关,因此这是和时间无关的Schrodinger方程。
,E是电子的基态能量。基态能量和时间无关,因此这是和时间无关的Schrodinger方程。
尽管电子波函数是所有N个电子坐标的函数,但仍可以将近似成单个电子波的乘机,即
![]() 。加入研究单原子维度还比较低,但是简单的CO2分子就有22相相乘,维度66维。研究有机物团簇那就更难以解决。所以实际的Schrodinger方程让许多顶尖学者困扰如此之久。
。加入研究单原子维度还比较低,但是简单的CO2分子就有22相相乘,维度66维。研究有机物团簇那就更难以解决。所以实际的Schrodinger方程让许多顶尖学者困扰如此之久。
除了电子波函数的维度问题,哈米尔顿量H,中关于电子-电子相互作用这一项在多原子求解时候相比于其他更为困难。
虽然求解Schrodinger是量子力学基本问题,但要明白并不能直接观测到基于某个体系的波函数。并且我们也并不关心每个电子的具体坐标,主要是关心整体的概率值。与此相关的物理量就是某个空间的具体位置上的电荷密度。可以用单电子波函数将其写成
![]()
此式中,针对电子系统占据的全部单电子波函数进行求和。因此,求和项中的表达式就是在单电子波函数![]() 中一个电子位于r处的概率值。表达式出现2是因为电子具有自旋。电荷密度
中一个电子位于r处的概率值。表达式出现2是因为电子具有自旋。电荷密度![]() ,仅仅是三维空间坐标的函数Schrodinger方程全波函数解的大量信息,计算这个比较简单,因为全波函数是3N个空间坐标的函数。
,仅仅是三维空间坐标的函数Schrodinger方程全波函数解的大量信息,计算这个比较简单,因为全波函数是3N个空间坐标的函数。
3密度泛函理论-从波函数到电荷密度
密度泛函理论是有Kohn,Hohenberg所证明的两个基本数学定理。第一个定理:从Schrodinger方程得到的基态能量是电荷密度的唯一函数。
定理表明:基态波函数和基态电荷密度之间,存在一一对应的关系。
泛函概念:函数的取值是由一个或者多个变量所决定,且由该变量确定了泛函的数值。一个简单的例子是单变量函数![]() 。泛函有点类似,但是其取值是由一个函数作为变量决定的,且该函数唯一的确定了泛函的数值。例如
。泛函有点类似,但是其取值是由一个函数作为变量决定的,且该函数唯一的确定了泛函的数值。例如
![]()
就是函数 的泛函。如果以
的泛函。如果以![]()
为例计算该泛函的值,就是![]() 。据此,可以将Hohenberg和Kohn结果重新定义表述为:基态能量E可以表达为E可以表达成
。据此,可以将Hohenberg和Kohn结果重新定义表述为:基态能量E可以表达为E可以表达成![]() ,其中
,其中![]() 是电荷密度这也就是为什么将这一理论称为密度泛函的原因。
是电荷密度这也就是为什么将这一理论称为密度泛函的原因。
另一个表述Hohenberg和Kohn结果的方式就是:基态电荷密度唯一的决定了基态的所有性质,包括能量和波函数。这意味着可以通过找到含有三个空间变量的电荷密度函数来求解Schrodinger方程,而不用求解3N个变量的波函数。
第一种形式虽然证明存在一个可以用来求解的Schrodinger方程的电荷密度泛函,但是该理论并没有给出求解该泛函的具体形式。第二个定理一个重要特征:使整体泛函最小化的电荷密度就是对应于schrodinger方程完全解的真实密度。如果已知这个真实的泛函形式,就能够通过不断调整电荷密度直到由泛函所确定的能量最小化,并可以找到相应的电荷密度。实际中,该变分原理常用于泛函的近似表达式。
将H-K定理描述的泛函写成单电子波函数![]() 的形式,是一个有益的办法。能量泛函:
的形式,是一个有益的办法。能量泛函:
![]()
将泛函写成简单解析形式的一项known,以及其他所有部分XC。known这一项包含电子的动能、电子和原子核之间的作用力、电子和电子之间的库伦作用力,原子核之间的库伦作用。XC一项是除了known之外的所有量子力学效应。
现在可以试想一下,我们可以采用更好的方式表达尚未确切定义的交换-关联能泛函。在寻找总能泛函最小的能量求解过程中,之前并没有很好的解决,后来Kohn和Sham解决了,给出这样一个结果:求解正确的电荷密度可以表示为求解一套方程,其中每一个方程都只和一个电子有关。
Kohn-Sham方程的表达式为:
相比于多体Schrodinger方程而言K-S方程取决于单电子波函数,少了求和。只取决于三个空间变量的单电子波函数。在K-S方程左侧,有三个势能项V、 \
\![]() 。第一项和Schrodinger定义相同,一个电子和所有原子核之间的相互作用。第二项也称为Hatree势能,可以写成
。第一项和Schrodinger定义相同,一个电子和所有原子核之间的相互作用。第二项也称为Hatree势能,可以写成
![]()
这个势能描述的是K-S方程考虑的单电子与全部电子所产生的电荷密度之间的库伦排斥作用。Hatree势能包含了一个自作用,因为一个电子本身也是总体电荷密度的一部分。所以在计算过程中总是包含了电子与自身的自作用的计算。但是问题在于,这个自作用在物理学中并不存在,是一个假想出来以便于表示和计算。所以还需要对此值进行修正,修正地部分放在![]() 中。但是这部分地修正只是
中。但是这部分地修正只是![]() 函数的一部分。
函数的一部分。![]() 在形式上面可以表示成交换关联能的“泛函导数”即
在形式上面可以表示成交换关联能的“泛函导数”即
![]()
与普通函数相比,这种形式并不容易理解。泛函法术的求导符号是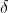 ,并不是d,这就是为了凸显他并不是一个普通的数学意义上的导数。
,并不是d,这就是为了凸显他并不是一个普通的数学意义上的导数。
在关于K-S方程的讨论中,似乎是陷入了一个循环。为了求解K-S方程,需要确定Hatree势能;为了得到Hatree势能,又需要知道电荷密度;为了找到电荷密度,又必须知道单电子波函数方程;为了知道这些波函数。又必须求解K-S方程。其实到这里迭代的初步思路已经展现,带入假设值一直进行迭代修正,最后输出一个误差在限制范围内的值。这就是K-S方程的自洽运算过程。