What‘s next for AlphaFold and the AI protein-folding revolution / 什么是AlphaFold和AI蛋白质折叠革命的下一步?
What's next for AlphaFold and the AI protein-folding revolution / 什么是AlphaFold和AI蛋白质折叠革命的下一步?
-
- DeepMind software that can predict the 3D shape of proteins is already changing biology.
- 可以预测蛋白质的三维结构的DeepMind 软件已经改变生物学
- A top-down view of the human nuclear pore complex, the largest molecular machine in human cells. Credit: Agnieszka Obarska-Kosinska
- 人类细胞核核孔复合体的俯视图,这是人类细胞中最大的分子机器。创作者: Agnieszka Obarska-Kosinska
- For more than a decade, molecular biologist Martin Beck and his colleagues have been trying to piece together one of the world’s hardest jigsaw puzzles: a detailed model of the largest molecular machine in human cells.
- 十多年来,人类分子生物学家 Martin Beck 和他的同事一直试图拼凑世界最大的拼图游戏之一:人类细胞分子机器详细模型。
- This behemoth, called the nuclear pore complex, controls the flow of molecules in and out of the nucleus of the cell, where the genome sits. Hundreds of these complexes exist in every cell. Each is made up of more than 1,000 proteins that together form rings around a hole through the nuclear membrane.
- 这个庞然大物,叫做核孔复合体,控制着进出细胞核的分子流,这里也是基因保存的地方,每个细胞包含有上百个复合体。每个由上千个蛋白质构成环绕成一个孔通过细胞核膜。
- These 1,000 puzzle pieces are drawn from more than 30 protein building blocks that interlace in myriad ways. Making the puzzle even harder, the experimentally determined 3D shapes of these building blocks are a potpourri of structures gathered from many species, so don’t always mesh together well. And the picture on the puzzle’s box — a low-resolution 3D view of the nuclear pore complex — lacks sufficient detail to know how many of the pieces precisely fit together.
- 这1,000 块拼图由 30 多种蛋白质构建块组成,这些蛋白质构建块以多种方式交织在一起。 使难题变得更加困难的是,这些构建块的实验确定的 3D 形状是从许多物种中收集的结构的综合,所以不是很好地融合在一起。 拼图盒子上的图片——核孔复合体的低分辨率 3D 视图——缺乏足够的细节来知道有多少部分精确地组合在一起。
- In 2016, a team led by Beck, who is based at the Max Planck Institute of Biophysics (MPIBP) in Frankfurt, Germany, reported a model1 that covered about 30% of the nuclear pore complex and around half of the 30 building blocks, called Nup proteins.
- 2016 年,由位于德国法兰克福马克斯普朗克生物物理研究所 (MPIBP) 的 Beck 领导的团队报告了一个模型 1,该模型覆盖了大约 30% 的核孔复合体和大约 30 个构建单元中的一半,称为 核蛋白。
- Then, last July, London-based firm DeepMind, part of Alphabet — Google’s parent company — made public an artificial intelligence (AI) tool called AlphaFold2. The software could predict the 3D shape of proteins from their genetic sequence with, for the most part, pinpoint accuracy. This transformed Beck’s task, and the studies of thousands of other biologists (see ‘AlphaFold mania’).
- 然后,去年 7 月,总部位于伦敦的 DeepMind 公司(谷歌母公司 Alphabet 的一部分)公开了一款名为 AlphaFold2 的人工智能 (AI) 工具。 该软件可以从蛋白质的基因序列中预测蛋白质的 3D 形状,并且在很大程度上具有精确度。 这改变了Beck 的任务,以及成千上万其他生物学家的研究(参见“AlphaFold 狂热”)。
- AlphaFold mania: bar chart that shows the number of research papers and preprints that have cited Alphafold since its release.
- AlphaFold 狂热:条形图显示自 Alphafold 发布以来引用的研究论文和预印本的数量。
- “AlphaFold changes the game,” says Beck. “This is like an earthquake. You can see it everywhere,” says Ora Schueler-Furman, a computational structural biologist at the Hebrew University of Jerusalem in Israel, who is using AlphaFold to model protein interactions. “There is before July and after.”
- “AlphaFold 改变了游戏规则,”贝克说。 “这就像一场地震。 你可以在任何地方看到它,”以色列耶路撒冷希伯来大学的计算结构生物学家 Ora Schueler-Furman 说,他正在使用 AlphaFold 来模拟蛋白质相互作用。 “七月之前和之后都有。”
- Using AlphaFold, Beck and others at the MPIBP — molecular biologist Agnieszka Obarska-Kosinska and a group led by biophysicist Gerhard Hummer — as well as a team led by structural modeller Jan Kosinski, at the European Molecular Biology Laboratory (EMBL) in Hamburg in Germany, could predict shapes for human versions of the Nup proteins more accurately. And by taking advantage of a tweak that helped AlphaFold to model how proteins interact, they managed to publish a model last October that covered 60% of the complex3. It reveals how the complex stabilizes holes in the nucleus, as well as hinting at how the complex controls what gets in and out.
- 使用 AlphaFold、Beck 和 MPIBP 的其他人——分子生物学家 Agnieszka Obarska-Kosinska 和由生物物理学家 Gerhard Hummer 领导的小组——以及由德国汉堡欧洲分子生物学实验室 (EMBL) 的结构建模师 Jan Kosinski 领导的小组 ,可以更准确地预测人类版本的 Nup 蛋白的形状。 通过利用帮助 AlphaFold 模拟蛋白质相互作用的调整,他们在去年 10 月成功发布了一个模型,涵盖了 60% 的复合体3。 它揭示了复合物如何稳定原子核中的孔,并暗示复合物如何控制进出的东西。
- DeepMind’s AI predicts structures for a vast trove of proteins
- DeepMind 的 AI 预测大量蛋白质的结构
- In the past half-year, AlphaFold mania has gripped the life sciences. “Every meeting I’m in, people are saying ‘why not use AlphaFold?’,” says Christine Orengo, a computational biologist at University College London.
- 在过去的半年里,AlphaFold 狂热席卷了生命科学领域。 “我参加的每次会议,人们都在说‘为什么不使用 AlphaFold?’,”伦敦大学学院的计算生物学家 Christine Orengo 说。
- In some cases, the AI has saved scientists time; in others it has made possible research that was previously inconceivable or wildly impractical. It has limitations, and some scientists are finding its predictions to be too unreliable for their work. But the pace of experimentation is frenetic.
- 在某些情况下,人工智能为科学家节省了时间; 在其他情况下,它使以前难以想象或非常不切实际的研究成为可能。 它有局限性,一些科学家发现它的预测对于他们的工作来说太不可靠了。 但实验的步伐是狂热的。
- Even those who developed the software are struggling to keep up with its use in areas ranging from drug discovery and protein design to the origins of complex life. “I wake up and type AlphaFold into Twitter,” says John Jumper, who leads the AlphaFold team at DeepMind. “It’s quite the experience to see everything.”
- 即使是开发该软件的人也在努力跟上它在从药物发现和蛋白质设计到复杂生命起源等领域的使用。 “我醒来并在 Twitter 上输入 AlphaFold,”领导 DeepMind AlphaFold 团队的 John Jumper 说。 “看到一切都是一种体验。”
- A startling success
- 惊人的成功
- AlphaFold caused a sensation in December 2020, when it dominated a contest called the Critical Assessment of Protein Structure Prediction, or CASP. The competition, held every two years, measures progress in one of biology’s grandest challenges: determining the 3D shapes of proteins from their amino-acid sequence alone. Computer-software entries are judged against structures of the same proteins determined using experimental methods such as X-ray crystallography or cryo-electron microscopy (cryo-EM), which fire X-rays or electron beams at proteins to build up a picture of their shape.
- AlphaFold 在 2020 年 12 月引起了轰动,当时它主导了一场名为“蛋白质结构预测关键评估”(CASP)的比赛。 该竞赛每两年举行一次,旨在衡量生物学最大挑战之一的进展:仅从蛋白质的氨基酸序列中确定蛋白质的 3D 形状。 计算机软件条目是根据使用 X 射线晶体学或低温电子显微镜 (cryo-EM) 等实验方法确定的相同蛋白质的结构来判断的,这些方法向蛋白质发射 X 射线或电子束以建立它们的图像 形状。
- The 2020 version of AlphaFold was the software’s second edition. It had also won the 2018 CASP, but its earlier efforts mostly weren’t good enough to stand in for experimentally determined structures, says Jumper. However, AlphaFold2’s predictions were, on average, on par with the empirical structures.
- AlphaFold 的 2020 版是该软件的第二版。 Jumper 说,它还赢得了 2018 年的 CASP,但其早期的努力大多不足以代替实验确定的结构。 然而,AlphaFold2 的预测平均而言与经验结构相当。
- It wasn’t clear when DeepMind would make the software or its predictions widely available, so researchers used information from a public talk by Jumper, and their own insights, to develop their own AI tool, called RoseTTAFold.
- 目前尚不清楚 DeepMind 何时会广泛使用该软件或其预测,因此研究人员利用 Jumper 公开演讲的信息和他们自己的见解,开发了自己的 AI 工具,称为 RoseTTAFold。
- Then on 15 July 2021, papers describing RoseTTAFold and AlphaFold2 appeared2,4, along with freely available, open-source code and other information needed for specialists to run their own versions of the tools. A week later, DeepMind announced that it had used AlphaFold to predict the structure of nearly every protein made by humans, as well as the entire ‘proteomes’ of 20 other widely studied organisms, such as mice and the bacterium Escherichia coli — more than 365,000 structures in total (see ‘What’s known about proteomes’). DeepMind also publicly released these to a database maintained by the EMBL’s European Bioinformatics Institute (EMBL–EBI), in Hinxton, UK. That database has since swelled to almost one million structures.
- 然后在 2021 年 7 月 15 日,出现了描述 RoseTTAFold 和 AlphaFold2 的论文2、4,以及免费提供的开源代码和专家运行他们自己的工具版本所需的其他信息。 一周后,DeepMind 宣布它已经使用 AlphaFold 预测了人类制造的几乎所有蛋白质的结构,以及其他 20 种广泛研究的生物体的整个“蛋白质组”,例如小鼠和大肠杆菌——超过 365,000 总结构(参见“关于蛋白质组的已知信息”)。 DeepMind 还将这些信息公开发布到由位于英国欣克斯顿的 EMBL 欧洲生物信息学研究所 (EMBL-EBI) 维护的数据库中。 此后,该数据库已膨胀到近一百万个结构。
- What’s known about proteomes: bar chart of percentage of structures from different species that come from PDB and AlphaFold.
- 关于蛋白质组的已知信息:来自 PDB 和 AlphaFold 的不同物种的结构百分比条形图。
- Source: E. Porta-Pardo et al. PLoS Comput. Biol. 18, e1009818 (2022).
- This year, DeepMind plans to release a total of more than 100 million structure predictions. That is nearly half of all known proteins — and hundreds of times more than the number of experimentally determined proteins in the Protein Data Bank (PDB) structure repository.
- 今年,DeepMind 计划发布总计超过 1 亿个结构预测。 这几乎是所有已知蛋白质的一半,是蛋白质数据库 (PDB) 结构库中实验确定的蛋白质数量的数百倍。
- AlphaFold deploys deep-learning neural networks: computational architectures inspired by the brain’s neural wiring to discern patterns in data. It has been trained on hundreds of thousands of experimentally determined protein structures and sequences in the PDB and other databases. Faced with a new sequence, it first looks for related sequences in databases, which can identify amino acids that have tended to evolve together, suggesting they’re close in 3D space. The structure of existing related proteins provides another way to estimate distances between amino-acid pairs in the new sequence.
- AlphaFold 部署了深度学习神经网络:受大脑神经线路启发的计算架构,可识别数据中的模式。 它已经接受了 PDB 和其他数据库中数十万个实验确定的蛋白质结构和序列的训练。 面对一个新序列,它首先在数据库中寻找相关序列,这些序列可以识别出倾向于一起进化的氨基酸,表明它们在 3D 空间中很接近。 现有相关蛋白质的结构提供了另一种估计新序列中氨基酸对之间距离的方法。
- AlphaFold iterates clues from these parallel tracks back and forth as it tries to model the 3D positions of amino acids, continually updating its estimate. Specialists say the software’s application of new ideas in machine learning research seems to be what makes AlphaFold so good — in particular, its use of an AI mechanism termed ‘attention’ to determine which amino-acid connections are most salient for its task at any moment.
- AlphaFold 在尝试对氨基酸的 3D 位置进行建模时来回迭代来自这些平行轨迹的线索,并不断更新其估计值。 专家表示,该软件在机器学习研究中的新思想应用似乎是 AlphaFold 如此出色的原因——特别是,它使用一种称为“注意力”的人工智能机制来确定哪些氨基酸连接在任何时候对其任务最重要 .
- DeepMind’s AI for protein structure is coming to the masses
- DeepMind 的蛋白质结构 AI 即将普及
- The network’s reliance on information about related protein sequences means that AlphaFold has some limitations. It is not designed to predict the effect of mutations, such as those that cause disease, on a protein’s shape. Nor was it trained to determine how proteins change shape in the presence of other interacting proteins, or molecules such as drugs. But its models come with scores that gauge the network’s confidence in its prediction for each amino-acid unit of a protein — and researchers are tweaking AlphaFold’s code to expand its capabilities.
- 该网络对相关蛋白质序列信息的依赖意味着 AlphaFold 存在一些局限性。 它并非旨在预测突变(例如导致疾病的突变)对蛋白质形状的影响。 它也没有被训练来确定在其他相互作用的蛋白质或药物等分子存在的情况下蛋白质如何改变形状。 但它的模型附带的分数可以衡量网络对其预测蛋白质每个氨基酸单元的信心——研究人员正在调整 AlphaFold 的代码以扩展其功能。
- By now, more than 400,000 people have used the EMBL-EBI’s AlphaFold database, according to DeepMind. There are also AlphaFold ‘power users’: researchers who’ve set up the software on their own servers or turned to cloud-based versions of AlphaFold to predict structures not in the EMBL-EBI database, or to dream up new uses for the tool.
- 据 DeepMind 称,到目前为止,已有超过 40 万人使用了 EMBL-EBI 的 AlphaFold 数据库。 还有 AlphaFold 的“超级用户”:研究人员在自己的服务器上安装了软件,或者转向基于云的 AlphaFold 版本来预测不在 EMBL-EBI 数据库中的结构,或者为该工具设想新用途 .
- Solving structures
- 求解结构
- Biologists are already impressed with AlphaFold’s ability to solve structures. “Based on what I’ve seen so far, I trust AlphaFold quite a lot,” says Thomas Boesen, a structural biologist at Aarhus University in Denmark. The software has successfully predicted the shapes of proteins that Boesen’s centre has determined but not yet published. “That’s a big validation from my side,” he says. He and Aarhus microbial ecologist Tina Šantl-Temkiv are using AlphaFold to model the structure of bacterial proteins that promote the formation of ice — and which could contribute to the cooling effects of ice in clouds — because biologists haven’t been able to fully determine the structures experimentally5.
- AlphaFold 解析结构的能力已经给生物学家留下了深刻的印象。 “根据我目前所见,我非常信任 AlphaFold,”丹麦奥胡斯大学的结构生物学家 Thomas Boesen 说。 该软件已成功预测了 Boesen 中心已确定但尚未发表的蛋白质形状。 “这对我来说是一个很大的验证,”他说。 他和奥胡斯微生物生态学家 Tina Šantl-Temkiv 正在使用 AlphaFold 来模拟细菌蛋白质的结构,这些蛋白质促进冰的形成——这可能有助于云中冰的冷却效应——因为生物学家还不能完全确定 结构实验5。
- As long as a protein curls up into a single well-defined 3D shape — and not all do — AlphaFold’s prediction can be hard to beat, says Arne Elofsson, a protein bioinformatician at Stockholm University. “It’s a one-click solution to get probably the best model you’re going to get.”
- 斯德哥尔摩大学的蛋白质生物信息学家 Arne Elofsson 说,只要一种蛋白质卷曲成一个定义明确的 3D 形状——而且并非全部如此——AlphaFold 的预测就很难被击败。 “这是一种一键式解决方案,可能是您将获得的最佳模型。”
- Where AlphaFold is less confident, “it’s very good at telling you when it doesn’t work”, Elofsson says. In such cases, predicted structures can resemble floating spaghetti strands (see ‘The good, the bad and the ugly’). This often corresponds to regions of proteins that lack a defined shape, at least in isolation. Such intrinsically disordered regions — which make up around one-third of the human proteome — might become well defined only when another molecule, such as a signalling partner, is present.
- Elofsson 说,在 AlphaFold 不太自信的地方,“它非常擅长告诉你什么时候它不起作用”。 在这种情况下,预测的结构可能类似于浮动的意大利面条线(参见“好的、坏的和丑陋的”)。 这通常对应于缺乏确定形状的蛋白质区域,至少在隔离时是这样。 这种本质上无序的区域——约占人类蛋白质组的三分之一——可能只有在存在另一种分子(如信号伙伴)时才能得到很好的定义。
- The good, the bad and the ugly: graphic that shows the varying accuracies of AlphaFold’s predictions with confidence estimates.
- 好的、坏的和丑陋的:图表显示了 AlphaFold 预测的不同准确性和置信度估计。
- Norman Davey, a computational biologist at the Institute of Cancer Research in London, says AlphaFold’s ability to identify disorder has been a game-changer for his work studying the properties of these regions. “Instantly there was a huge increase in the quality of the predictions we had, without any effort on our part,” he says.
- 伦敦癌症研究所的计算生物学家 Norman Davey 表示,AlphaFold 识别疾病的能力已经改变了他研究这些区域特性的工作。 他说:“我们的预测质量立即有了巨大的提高,而我们没有付出任何努力。”
- AlphaFold’s dump of protein structures into the EMBL-EBI database is also immediately being put to use. Orengo’s team is searching it to identify fresh kinds of proteins (without experimentally verifying them) and has turned up hundreds, perhaps thousands, of potentially new protein families, expanding scientists’ knowledge of what proteins look like and can do. In another effort, the team is scouring databases of DNA sequences harvested from the ocean and waste water, to try to identify new plastic-eating enzymes. Using AlphaFold to quickly approximate the structures of thousands of proteins, the researchers hope to better understand how enzymes evolved to break down plastic, and potentially to improve them.
- AlphaFold 将蛋白质结构转储到 EMBL-EBI 数据库中的数据也立即投入使用。 Orengo 的团队正在搜索它以识别新的蛋白质种类(没有通过实验验证它们),并且已经发现了数百甚至数千个潜在的新蛋白质家族,扩大了科学家对蛋白质外观和功能的了解。 在另一项努力中,该团队正在搜索从海洋和废水中采集的 DNA 序列数据库,以尝试识别新的食用塑料酶。 使用 AlphaFold 快速近似数千种蛋白质的结构,研究人员希望更好地了解酶如何进化以分解塑料,并有可能改进它们。
- The ability to transform any protein-coding gene sequence into a reliable structure should be especially powerful for evolution studies, says Sergey Ovchinnikov, an evolutionary biologist at Harvard University in Cambridge, Massachusetts. Researchers compare genetic sequences to determine how organisms and their genes are related across species. For distantly related genes, comparisons might fail to turn up evolutionary relatives because the sequences have changed so much. But by comparing protein structures — which tend to change less rapidly than genetic sequences — researchers might be able to uncover overlooked ancient relationships. “This opens up an amazing opportunity to study the evolution of proteins and the origins of life,” says Pedro Beltrao, a computational biologist at the Swiss Federal Institute of Technology in Zurich.
- 马萨诸塞州剑桥市哈佛大学的进化生物学家 Sergey Ovchinnikov 说,将任何蛋白质编码基因序列转化为可靠结构的能力对于进化研究来说应该是特别强大的。 研究人员比较基因序列以确定生物及其基因在物种间的相关性。 对于远缘相关的基因,比较可能无法找到进化亲属,因为序列发生了很大变化。 但通过比较蛋白质结构——其变化往往不如基因序列快——研究人员或许能够发现被忽视的古老关系。 苏黎世瑞士联邦理工学院的计算生物学家 Pedro Beltrao 说:“这为研究蛋白质进化和生命起源提供了一个绝佳的机会。”
- To test this idea, a team led by Martin Steinegger, a computational biologist at Seoul National University, and his colleagues used a tool they developed, called Foldseek, to look for relatives of the RNA-copying enzyme of SARS-CoV-2 — the virus that causes COVID-19 — in the EMBL-EBI’s AlphaFold database6. This search turned up previously unidentified possible ancient relatives: proteins across eukaryotes — including slime moulds — that resemble, in their 3D structure, enzymes called reverse transcriptases that viruses such as HIV use to copy RNA into DNA, despite very little similarity at the genetic-sequence level.
- 为了验证这个想法,首尔国立大学的计算生物学家 Martin Steinegger 和他的同事领导的一个团队使用他们开发的名为 Foldseek 的工具来寻找 SARS-CoV-2 的 RNA 复制酶的亲属—— 导致 COVID-19 的病毒——在 EMBL-EBI 的 AlphaFold 数据库中6。 这项搜索发现了以前未知的可能的远古亲属:真核生物中的蛋白质——包括粘菌——在它们的 3D 结构中类似于称为逆转录酶的酶,病毒如 HIV 使用逆转录酶将 RNA 复制到 DNA 中,尽管在遗传上几乎没有相似性。 序列级别。
- For scientists who want to determine the detailed structure of a specific protein, an AlphaFold prediction isn’t necessarily an immediate solution. Rather, it provides an initial approximation that can be validated or refined by experiment — and which itself helps to make sense of experimental data. Raw data from X-ray crystallography, for instance, appear as patterns of diffracted X-rays. Typically, scientists need a starting guess at a protein’s structure to interpret these patterns. Previously, they’d often cobble together information from related proteins in the PDB or use experimental approaches, says Randy Read, a structural biologist at the University of Cambridge, UK, whose lab specialized in some of these methods. Now, AlphaFold’s predictions have rendered such approaches unnecessary for most X-ray patterns, Read says, and his lab is working to make better use of AlphaFold in experimental models. “We’ve totally refocused our research.”
- 对于想要确定特定蛋白质的详细结构的科学家来说,AlphaFold 预测不一定是立竿见影的解决方案。 相反,它提供了一个可以通过实验验证或改进的初始近似值——它本身有助于理解实验数据。 例如,来自 X 射线晶体学的原始数据显示为衍射 X 射线的图案。 通常,科学家需要对蛋白质结构进行初步猜测才能解释这些模式。 英国剑桥大学的结构生物学家 Randy Read 说,以前,他们经常将 PDB 中相关蛋白质的信息拼凑在一起,或者使用实验方法。他的实验室专门研究其中一些方法。 现在,AlphaFold 的预测使得大多数 X 射线模式不需要这种方法,Read 说,他的实验室正在努力在实验模型中更好地利用 AlphaFold。 “我们完全重新调整了研究重点。”
- Artificial intelligence powers protein-folding predictions
-
- 人工智能为蛋白质折叠预测提供动力
- He and other researchers have used AlphaFold to determine crystal structures from X-ray data that were uninterpretable without an adequate starting model. “People are solving structures that, for years, had not been solved,” says Claudia Millán Nebot, a former postdoc in Read’s lab who now works at the analytics firm SciBite in Cambridge. She expects to see a glut of new protein structures submitted to the PDB, in large part as a result of AlphaFold.
- 他和其他研究人员已经使用 AlphaFold 从 X 射线数据中确定晶体结构,这些数据在没有足够的起始模型的情况下是无法解释的。 “人们正在解决多年来一直没有解决的结构,”Claudia Millán Nebot 说,他是 Read 实验室的前博士后,现在在剑桥的分析公司 SciBite 工作。 她预计会看到大量新的蛋白质结构提交给 PDB,这在很大程度上是 AlphaFold 的结果。
- The same is true for labs specializing in cryo-EM, which captures pictures of flash-frozen proteins. In some instances, AlphaFold’s models have accurately predicted unique features of proteins called G-protein-coupled receptors (GPCRs) — which are important drug targets — that other computational tools got wrong, says Bryan Roth, a structural biologist and pharmacologist at the University of North Carolina at Chapel Hill. “It seems to be really good for generating first models, which we then refine with some experimental data,” he says. “That saves us some time.”
- 专门从事冷冻电镜研究的实验室也是如此,它可以捕捉快速冷冻蛋白质的照片。 在某些情况下,AlphaFold 的模型准确地预测了称为 G 蛋白偶联受体 (GPCR) 的蛋白质的独特特征,这些蛋白质是重要的药物靶标,而其他计算工具出错了,美国大学的结构生物学家和药理学家 Bryan Roth 说 北卡罗来纳州教堂山。 “它似乎非常适合生成第一个模型,然后我们用一些实验数据对其进行改进,”他说。 “这为我们节省了一些时间。”
- But Roth adds that AlphaFold isn’t always that accurate. Of the several dozen GPCR structures his lab has solved, but not yet published, he says, “about half the time, the AlphaFold structures are fairly good, and half the time they’re more or less useless for our purposes”. In some instances, he says, AlphaFold labels predictions with high confidence, but experimental structures show that it is wrong. Even when the software gets it right, it cannot model how a protein would look when bound to a drug or other small molecule (ligand), which can substantially alter the structure. Such caveats make Roth wonder how useful AlphaFold will be for drug discovery.
- 但 Roth 补充说,AlphaFold 并不总是那么准确。 他说,在他的实验室已经解决但尚未发表的几十个 GPCR 结构中,“大约有一半的时间,AlphaFold 结构相当好,而有一半的时间它们或多或少对我们的目的毫无用处”。 他说,在某些情况下,AlphaFold 以高置信度标记预测,但实验结构表明它是错误的。 即使软件做对了,它也无法模拟蛋白质与药物或其他小分子(配体)结合时的外观,这会大大改变结构。 这些警告让 Roth 想知道 AlphaFold 对药物发现有多大用处。
- It’s increasingly common in drug-discovery efforts to use computational-docking software that screens billions of small molecules to find some that might bind to proteins — one indication that they could make useful drugs. Roth is now working with Brian Shoichet, a medicinal chemist at the University of California, San Francisco, to see how AlphaFold’s predictions compare with experimentally determined structures in this exercise.
- 在药物发现工作中,使用计算对接软件越来越普遍,该软件可以筛选数十亿个小分子,以找到一些可能与蛋白质结合的分子——这表明它们可以制造有用的药物。 Roth 现在正与加州大学旧金山分校的药物化学家 Brian Shoichet 合作,以了解 AlphaFold 的预测如何与本练习中通过实验确定的结构进行比较。
- Shoichet says they are limiting their work to proteins for which AlphaFold’s prediction chimes with experimental structures. But even in these instances, the docking software is turning up different drug hits for the experimental structure and AlphaFold’s take, suggesting that small discrepancies could matter. “That doesn’t mean we won’t find new ligands, we’ll just find different ones,” says Shoichet. His team is now synthesizing potential drugs identified using AlphaFold structures, and testing their activity in the lab.
- Shoichet 说,他们将工作限制在 AlphaFold 的预测与实验结构相吻合的蛋白质上。 但即使在这些情况下,对接软件也会为实验结构和 AlphaFold 提供不同的药物命中率,这表明微小的差异可能很重要。 “这并不意味着我们不会找到新的配体,我们只会找到不同的配体,”Shoichet 说。 他的团队现在正在合成使用 AlphaFold 结构识别的潜在药物,并在实验室中测试它们的活性。
- Critical optimism
-
- Researchers at pharmaceutical companies and biotechnology firms are excited about AlphaFold’s potential to help with drug discovery, says Shoichet. “Critical optimism is how I’d describe it.” In November 2021, DeepMind launched its own spin-off, Isomorphic Labs, which aims to apply AlphaFold and other AI tools to drug discovery. But the company has said little else about its plans.
- Shoichet 说,制药公司和生物技术公司的研究人员对 AlphaFold 帮助药物发现的潜力感到兴奋。 “批判性的乐观是我描述它的方式。” 2021 年 11 月,DeepMind 推出了自己的衍生产品 Isomorphic Labs,旨在将 AlphaFold 和其他 AI 工具应用于药物发现。 但该公司对其计划只字未提。
- Karen Akinsanya, who leads therapeutics development at Schrödinger, a drug-discovery firm headquartered in New York City that also publishes chemical-simulations software, says she and her colleagues are already having some success using AlphaFold structures, including for GPCRs, in virtual screens and compound design for drug candidates. She finds that, just as with experimental structures, extra software is needed to get at the fine details of amino-acid side chains or locations where individual hydrogen atoms might sit. Once this is done, AlphaFold structures have proved good enough to guide drug discovery — in some cases.
- Karen Akinsanya 是 Schrödinger 的治疗开发负责人,Schrödinger 是一家总部位于纽约市的药物发现公司,还发布了化学模拟软件,她说她和她的同事已经在虚拟屏幕和 GPCR 中使用 AlphaFold 结构取得了一些成功。 候选药物的化合物设计。 她发现,就像实验结构一样,需要额外的软件来获取氨基酸侧链或单个氢原子可能所在位置的详细信息。 一旦完成,AlphaFold 结构已被证明足以指导药物发现——在某些情况下。
- “It’s hard to say ‘this is a panacea’; that because you can do it very well for one structure — surprisingly and excitingly well — that it is eminently applicable to all structures. It clearly isn’t,” Akinsanya says. And she and her colleagues have found that AlphaFold’s accuracy predictions don’t show whether a structure will be useful for later drug screening. AlphaFold structures will never fully replace experimental ones in drug discovery, she says. But they might speed up the process by complementing experimental methods.
- “很难说‘这是灵丹妙药’; 因为你可以为一个结构做得很好——令人惊讶和令人兴奋的——它非常适用于所有结构。 显然不是,”Akinsanya 说。 她和她的同事发现,AlphaFold 的准确性预测并不能显示一个结构是否对以后的药物筛选有用。 她说,AlphaFold 结构永远不会完全取代药物发现中的实验性结构。 但他们可能会通过补充实验方法来加速这一过程。
- Drug developers curious about AlphaFold received good news in January, when DeepMind lifted a key restriction on its use for commercial applications. When the company released AlphaFold’s code in July 2021, it had stipulated that the parameters, or weights, needed to run the AlphaFold neural network — the end result of training the network on hundreds of thousands of protein structures and sequences — were for non-commercial use only. Akinsanya says this was a bottleneck for some in industry, and there was a “wave of excitement” when DeepMind changed tack. (RoseTTAFold came with similar restrictions, says Ovchinnikov, one of its developers. But the next version will be fully open-source.)
- 对 AlphaFold 感到好奇的药物开发人员在 1 月份收到了好消息,当时 DeepMind 取消了对其用于商业应用的关键限制。 当该公司在 2021 年 7 月发布 AlphaFold 的代码时,它规定运行 AlphaFold 神经网络所需的参数或权重——这是在数十万个蛋白质结构和序列上训练网络的最终结果——用于非商业用途 仅使用。 Akinsanya 说,这对行业中的一些人来说是一个瓶颈,当 DeepMind 改变策略时出现了一股“兴奋的浪潮”。 (RoseTTAFold 也有类似的限制,其开发人员之一 Ovchinnikov 说。但下一个版本将完全开源。)
- AI tools are not just changing how scientists determine what proteins look like. Some researchers are using them to make entirely new proteins. “Deep learning is completely transforming the way that protein design is being done in my group,” says David Baker, a biochemist at the University of Washington in Seattle and a leader in the field of designing proteins, as well as predicting their structures. His team, with computational chemist Minkyung Baek, led the work to develop RoseTTAFold.
- 人工智能工具不仅改变了科学家确定蛋白质外观的方式。 一些研究人员正在使用它们来制造全新的蛋白质。 “深度学习正在彻底改变我团队中蛋白质设计的方式,”西雅图华盛顿大学的生物化学家、蛋白质设计和预测其结构领域的领导者大卫贝克说。 他的团队与计算化学家 Minkyung Baek 一起领导了开发 RoseTTAFold 的工作。
- Baker’s team gets AlphaFold and RoseTTAFold to “hallucinate” new proteins. The researchers have altered the AI code so that, given random sequences of amino acids, the software will optimize them until they resemble something that the neural networks recognize as a protein (see ‘Dreaming up proteins’).
- Baker 的团队让 AlphaFold 和 RoseTTAFold 能够“产生幻觉”新的蛋白质。 研究人员已经改变了人工智能代码,因此,给定氨基酸的随机序列,软件将对其进行优化,直到它们类似于神经网络识别为蛋白质的东西(参见“梦想蛋白质”)。
- Dreaming up proteins: graphic that compares a protein structure predicted by a neural network with an actual stucture.
- 梦想蛋白质:将神经网络预测的蛋白质结构与实际结构进行比较的图形。
-
- In December 2021, Baker and his colleagues reported expressing 129 of these hallucinated proteins in bacteria, and found that about one-fifth of them folded into something resembling their predicted shape7. “That’s really the first demonstration that you can design proteins using these networks,” Baker says. His team is now using this approach to design proteins that do useful things, such as catalyse a particular chemical reaction, by specifying the amino acids responsible for the desired function and letting the AI dream up the rest.
- 2021 年 12 月,贝克和他的同事报告说,在细菌中表达了 129 种这些幻觉蛋白,并发现其中约五分之一折叠成类似于其预测形状的东西 7。 “这确实是第一次证明你可以使用这些网络设计蛋白质,”贝克说。 他的团队现在正在使用这种方法来设计做有用事情的蛋白质,例如催化特定的化学反应,方法是指定负责所需功能的氨基酸,并让 AI 梦想其余部分。
- Animation of four protein structures being predicted by the Alphafold AI system
- Alphafold AI 系统预测的四种蛋白质结构的动画
-
- Four examples of protein ‘hallucination’. In each case, AlphaFold is presented with a random amino-acid sequence, predicts the structure, and changes the sequence until the software confidently predicts that it will fold into a protein with a well-defined 3D shape. Colours show prediction confidence (from red for very low confidence, through yellow and light blue to dark blue for very high confidence). Initial frames have been slowed down for clarity.Credit: Sergey Ovchinnikov
- 蛋白质“幻觉”的四个例子。 在每种情况下,AlphaFold 都会显示一个随机氨基酸序列,预测结构并更改序列,直到软件有把握地预测它将折叠成具有明确 3D 形状的蛋白质。 颜色显示预测置信度(从红色表示非常低的置信度,通过黄色和浅蓝色到深蓝色表示非常高的置信度)。 为了清晰起见,最初的帧已经放慢了。Credit: Sergey Ovchinnikov
- Hacking AlphaFold
-
- When DeepMind released its AlphaFold code, Ovchinnikov wanted to better understand how the tool worked. Within days, he and computational-biology colleagues, including Steinegger, set up a website called ColabFold that allowed anyone to submit a protein sequence to AlphaFold or RoseTTAFold and get a structure prediction. Ovchinnikov imagined that he and other scientists would use ColabFold to try and ‘break’ AlphaFold, for instance, by supplying false information about a target protein sequence’s evolutionary relatives. By doing this, Ovchinnikov hoped he could determine how the network had learnt to predict structures so well.
- 当 DeepMind 发布其 AlphaFold 代码时,Ovchinnikov 想要更好地了解该工具的工作原理。 几天之内,他和包括 Steinegger 在内的计算生物学同事建立了一个名为 ColabFold 的网站,允许任何人向 AlphaFold 或 RoseTTAFold 提交蛋白质序列并获得结构预测。 Ovchinnikov 设想他和其他科学家会使用 ColabFold 来尝试“破坏”AlphaFold,例如,通过提供有关目标蛋白质序列进化亲属的虚假信息。 通过这样做,Ovchinnikov 希望他能够确定网络是如何学会如此出色地预测结构的。
- As it turned out, most researchers who used ColabFold just wanted to get a protein structure. But others used it as a platform to modify the inputs to AlphaFold to tackle new applications. “I didn’t expect the number of hacks of various types,” says Jumper.
- 事实证明,大多数使用 ColabFold 的研究人员只是想获得蛋白质结构。 但其他人将其用作修改 AlphaFold 的输入以处理新应用程序的平台。 “我没想到会出现各种类型的黑客攻击,”Jumper 说。
- By far the most popular hack has been to wield the tool on protein complexes comprised of multiple, interacting — and often intertwined — chains of peptides. Just as with the nuclear pore complex, many proteins in cells gain their function when they form complexes with multiple protein subunits.
- 到目前为止,最流行的黑客攻击是在蛋白质复合物上使用该工具,该复合物由多个相互作用的——通常是相互交织的——肽链组成。 就像核孔复合物一样,细胞中的许多蛋白质在与多个蛋白质亚基形成复合物时发挥作用。
- AlphaFold was designed to predict the shape of single peptide chains, and its training consisted entirely of such proteins. But the network seems to have learnt something about how complexes fold together. Several days after AlphaFold’s code was released, Yoshitaka Moriwaki, a protein bioinformatician at the University of Tokyo, tweeted that it could accurately predict interactions between two protein sequences if they were stitched together with a long linker sequence. Baek soon shared another hack to predict complexes, gleaned from developing RoseTTAFold.
- AlphaFold 旨在预测单个肽链的形状,其训练完全由此类蛋白质组成。 但该网络似乎已经了解了一些关于复合物如何折叠在一起的知识。 AlphaFold 的代码发布几天后,东京大学的蛋白质生物信息学家 Yoshitaka Moriwaki 在推特上表示,如果将两个蛋白质序列与长连接序列缝合在一起,它可以准确预测它们之间的相互作用。 Baek 很快分享了另一个从开发 RoseTTAFold 中收集到的预测复合物的技巧。
- ColabFold later incorporated the ability to predict complexes. And in October 2021, DeepMind released an update called AlphaFold-Multimer8 that was specifically trained on protein complexes, unlike its predecessor. Jumper’s team applied it to thousands of complexes in the PDB, and found that it predicted around 70% of the known protein–protein interactions.
- ColabFold 后来加入了预测复合物的能力。 并且在 2021 年 10 月,DeepMind 发布了一个名为 AlphaFold-Multimer8 的更新,该更新专门针对蛋白质复合物进行训练,与其前身不同。 Jumper 的团队将其应用于 PDB 中的数千个复合物,发现它预测了大约 70% 的已知蛋白质-蛋白质相互作用。
- These tools are already helping researchers to spot potential new protein partners. Elofsson’s team used AlphaFold to predict the structures of 65,000 human protein pairs that were suspected to interact on the basis of experimental data9. And a team led by Baker used AlphaFold and RoseTTAFold to model interactions between nearly every pair of proteins encoded by yeast, identifying more than 100 previously unknown complexes10. Such screens are just starting points, says Elofsson. They do a good job of predicting some protein pairings, particularly those that are stable, but struggle to identify more transient interactions. “Because it looks nice doesn’t mean it is correct,” says Elofsson. “You need some experimental data that show you’re right.”
- 这些工具已经在帮助研究人员发现潜在的新蛋白质伙伴。 Elofsson 的团队使用 AlphaFold 预测了 65,000 个人类蛋白质对的结构,这些蛋白质对根据实验数据被怀疑相互作用9。 Baker 领导的一个团队使用 AlphaFold 和 RoseTTAFold 来模拟酵母编码的几乎每一对蛋白质之间的相互作用,识别出 100 多个以前未知的复合物 10。 Elofsson 说,这样的屏幕只是起点。 他们在预测某些蛋白质配对方面做得很好,尤其是那些稳定但难以识别更多瞬时相互作用的蛋白质配对。 “因为它看起来不错并不意味着它是正确的,”Elofsson 说。 “你需要一些实验数据来证明你是对的。”
- The nuclear pore complex work is a good example of how predictions and experimental data can work together, says Kosinski (see ‘Genome gateway’). “It’s not like we take all the 30 proteins, throw them into AlphaFold and get the structure out.” To put the predicted protein structures together, the team used 3D images of the nuclear pore complex, captured using a form of cryo-EM called cryo-electron tomography. In one instance, experiments that can determine the proximity of proteins turned up a surprising interaction between two components of the complex, which AlphaFold’s models then confirmed.
- Kosinski 说,核孔复合体工作是预测和实验数据如何协同工作的一个很好的例子(参见“基因组网关”)。 “这并不是说我们将所有 30 种蛋白质都放入 AlphaFold 中并取出结构。” 为了将预测的蛋白质结构组合在一起,该团队使用了核孔复合物的 3D 图像,这些图像是使用一种称为低温电子断层扫描的低温电子显微镜拍摄的。 在一个例子中,可以确定蛋白质接近度的实验在复合物的两个成分之间产生了令人惊讶的相互作用,AlphaFold 的模型随后证实了这一点。
- Genome gateway: Two views of the human nuclear pore complex showing how it embeds in the nuclear membrane
- 基因组网关:人类核孔复合体的两种视图显示它如何嵌入核膜
- Images adapted from ref. 3/Agnieszka Obarska-Kosinska
-
- Kosinski sees the team’s current map of the nuclear pore complex as a starting point for experiments and simulations that examine how the pore complex functions — and how it malfunctions in disease.
- Kosinski 将团队当前的核孔复合体地图视为实验和模拟的起点,这些实验和模拟检查了孔复合体的功能 - 以及它如何在疾病中出现故障。
- AlphaFold’s limits
-
- For all the progress made with AlphaFold, scientists say that it is important to be clear about its limitations — particularly because researchers who don’t specialize in predicting protein structures use it.
- 对于 AlphaFold 取得的所有进展,科学家们表示,重要的是要清楚它的局限性——特别是因为不专门预测蛋白质结构的研究人员会使用它。
- Attempts to apply AlphaFold to various mutations that disrupt a protein’s natural structure, including one linked to early breast cancer, have confirmed that the software is not equipped to predict the consequences of new mutations in proteins, since there are no evolutionarily-related sequences to examine11.
- 尝试将 AlphaFold 应用于破坏蛋白质自然结构的各种突变,包括与早期乳腺癌相关的突变,已证实该软件无法预测蛋白质新突变的后果,因为没有进化相关的序列可供检查11 .
- The AlphaFold team is now thinking about how a neural network could be designed to deal with new mutations. Jumper expects this would require the network to better predict how a protein goes from its unfolded to its folded state. That would probably need software that relies only on what it has learnt about protein physics to predict structures, says Mohammed AlQuraishi, a computational biologist at Columbia University in New York City. “One thing we are interested in is making predictions from single sequences without using evolutionary information,” he says. “That’s a key problem that does remain open.”
- AlphaFold 团队现在正在考虑如何设计神经网络来处理新的突变。 Jumper 预计这将需要网络更好地预测蛋白质如何从展开状态变为折叠状态。 纽约市哥伦比亚大学的计算生物学家 Mohammed AlQuraishi 说,这可能需要仅依靠它所学到的蛋白质物理学知识来预测结构的软件。 “我们感兴趣的一件事是在不使用进化信息的情况下从单个序列进行预测,”他说。 “这是一个尚未解决的关键问题。”
- AlphaFold is also designed to predict a single structure, although it has been hacked to spit out more than one. But many proteins take on multiple conformations, which can be important to their function. “AlphaFold can’t really deal with proteins that can adopt different structures in different conformations,” says Schueler-Furman. And the predictions are for structures in isolation, whereas many proteins function alongside ligands such as DNA and RNA, fat molecules and minerals such as iron. “We are still missing ligands, we are missing everything else about proteins,” says Elofsson.
- AlphaFold 也被设计用来预测一个单一的结构,尽管它已经被黑客破解了不止一个。 但是许多蛋白质具有多种构象,这对其功能可能很重要。 “AlphaFold 不能真正处理可以采用不同构象的不同结构的蛋白质,”Schueler-Furman 说。 并且预测是针对孤立结构的,而许多蛋白质与配体(如 DNA 和 RNA)、脂肪分子和矿物质(如铁)一起发挥作用。 “我们仍然缺少配体,我们缺少关于蛋白质的其他一切,”Elofsson 说。
- Developing these next-generation neural networks will be a huge challenge, says AlQuraishi. AlphaFold relied on decades of research which generated experimental structures of proteins that the network could learn from. That volume of data is currently not available to capture protein dynamics, or the shapes of the trillions of smaller molecules that proteins could interact with. The PDB includes structures of proteins as they interact with other molecules, but this captures just a sliver of chemical diversity, Jumper adds.
- AlQuraishi 说,开发这些下一代神经网络将是一个巨大的挑战。 AlphaFold 依赖于数十年的研究,这些研究产生了网络可以学习的蛋白质实验结构。 目前无法获得如此大量的数据来捕捉蛋白质动力学,或者蛋白质可以与之相互作用的数万亿个小分子的形状。 Jumper 补充说,PDB 包括蛋白质与其他分子相互作用时的结构,但这仅捕获了一小部分化学多样性。
- Researchers think that it will take time for them to determine how best to wield AlphaFold and related AI tools. AlQuraishi sees parallels with the early days of television, when some programmes consisted of radio broadcasters simply reading the news. “I think we’re going to find new applications of structure that we haven’t conceived of yet.”
- 研究人员认为,他们需要时间来确定如何最好地使用 AlphaFold 和相关的人工智能工具。 AlQuraishi 看到了电视早期的相似之处,当时一些节目由广播电台组成,只是阅读新闻。 “我认为我们将找到我们尚未想到的结构的新应用。”
- Where the AlphaFold revolution is ends up is anybody’s guess. “Things are just changing so fast,” says Baker. “Even in the next year, we’re going to see really major breakthroughs made using these tools.” Janet Thornton, a computational biologist at the EMBL-EBI, thinks one of AlphaFold’s biggest impacts might be simply to convince biologists to be more open to insights from computational and theoretical approaches. “To me, the revolution is the mindset change,” she says.
- AlphaFold 革命的终点在哪里,谁也说不准。 “事情变化太快了,”贝克说。 “即使在明年,我们也将看到使用这些工具取得的重大突破。” EMBL-EBI 的计算生物学家 Janet Thornton 认为,AlphaFold 的最大影响之一可能只是说服生物学家对计算和理论方法的见解更加开放。 “对我来说,革命就是思维方式的改变,”她说。
- The AlphaFold revolution has inspired Kosinski to dream big. He imagines that AlphaFold-inspired tools could be used to model not just individual proteins and complexes, but entire organelles or even cells down to the level of individual protein molecules. “This is the dream we will follow for the next decades.”
- AlphaFold 革命激发了 Kosinski 的远大梦想。 他认为受 AlphaFold 启发的工具不仅可用于对单个蛋白质和复合物进行建模,还可以对整个细胞器甚至细胞进行建模,直至单个蛋白质分子的水平。 “这是我们未来几十年的梦想。”
- References
DeepMind software that can predict the 3D shape of proteins is already changing biology.
可以预测蛋白质的三维结构的DeepMind 软件已经改变生物学
A top-down view of the human nuclear pore complex, the largest molecular machine in human cells. Credit: Agnieszka Obarska-Kosinska
人类细胞核核孔复合体的俯视图,这是人类细胞中最大的分子机器。创作者: Agnieszka Obarska-Kosinska
For more than a decade, molecular biologist Martin Beck and his colleagues have been trying to piece together one of the world’s hardest jigsaw puzzles: a detailed model of the largest molecular machine in human cells.
十多年来,人类分子生物学家 Martin Beck 和他的同事一直试图拼凑世界最大的拼图游戏之一:人类细胞分子机器详细模型。
This behemoth, called the nuclear pore complex, controls the flow of molecules in and out of the nucleus of the cell, where the genome sits. Hundreds of these complexes exist in every cell. Each is made up of more than 1,000 proteins that together form rings around a hole through the nuclear membrane.
这个庞然大物,叫做核孔复合体,控制着进出细胞核的分子流,这里也是基因保存的地方,每个细胞包含有上百个复合体。每个由上千个蛋白质构成环绕成一个孔通过细胞核膜。
These 1,000 puzzle pieces are drawn from more than 30 protein building blocks that interlace in myriad ways. Making the puzzle even harder, the experimentally determined 3D shapes of these building blocks are a potpourri of structures gathered from many species, so don’t always mesh together well. And the picture on the puzzle’s box — a low-resolution 3D view of the nuclear pore complex — lacks sufficient detail to know how many of the pieces precisely fit together.
这1,000 块拼图由 30 多种蛋白质构建块组成,这些蛋白质构建块以多种方式交织在一起。 使难题变得更加困难的是,这些构建块的实验确定的 3D 形状是从许多物种中收集的结构的综合,所以不是很好地融合在一起。 拼图盒子上的图片——核孔复合体的低分辨率 3D 视图——缺乏足够的细节来知道有多少部分精确地组合在一起。
In 2016, a team led by Beck, who is based at the Max Planck Institute of Biophysics (MPIBP) in Frankfurt, Germany, reported a model1 that covered about 30% of the nuclear pore complex and around half of the 30 building blocks, called Nup proteins.
2016 年,由位于德国法兰克福马克斯普朗克生物物理研究所 (MPIBP) 的 Beck 领导的团队报告了一个模型 1,该模型覆盖了大约 30% 的核孔复合体和大约 30 个构建单元中的一半,称为 核蛋白。
Then, last July, London-based firm DeepMind, part of Alphabet — Google’s parent company — made public an artificial intelligence (AI) tool called AlphaFold2. The software could predict the 3D shape of proteins from their genetic sequence with, for the most part, pinpoint accuracy. This transformed Beck’s task, and the studies of thousands of other biologists (see ‘AlphaFold mania’).
然后,去年 7 月,总部位于伦敦的 DeepMind 公司(谷歌母公司 Alphabet 的一部分)公开了一款名为 AlphaFold2 的人工智能 (AI) 工具。 该软件可以从蛋白质的基因序列中预测蛋白质的 3D 形状,并且在很大程度上具有精确度。 这改变了Beck 的任务,以及成千上万其他生物学家的研究(参见“AlphaFold 狂热”)。

AlphaFold mania: bar chart that shows the number of research papers and preprints that have cited Alphafold since its release.
AlphaFold 狂热:条形图显示自 Alphafold 发布以来引用的研究论文和预印本的数量。
“AlphaFold changes the game,” says Beck. “This is like an earthquake. You can see it everywhere,” says Ora Schueler-Furman, a computational structural biologist at the Hebrew University of Jerusalem in Israel, who is using AlphaFold to model protein interactions. “There is before July and after.”
“AlphaFold 改变了游戏规则,”贝克说。 “这就像一场地震。 你可以在任何地方看到它,”以色列耶路撒冷希伯来大学的计算结构生物学家 Ora Schueler-Furman 说,他正在使用 AlphaFold 来模拟蛋白质相互作用。 “七月之前和之后都有。”
Using AlphaFold, Beck and others at the MPIBP — molecular biologist Agnieszka Obarska-Kosinska and a group led by biophysicist Gerhard Hummer — as well as a team led by structural modeller Jan Kosinski, at the European Molecular Biology Laboratory (EMBL) in Hamburg in Germany, could predict shapes for human versions of the Nup proteins more accurately. And by taking advantage of a tweak that helped AlphaFold to model how proteins interact, they managed to publish a model last October that covered 60% of the complex3. It reveals how the complex stabilizes holes in the nucleus, as well as hinting at how the complex controls what gets in and out.
使用 AlphaFold、Beck 和 MPIBP 的其他人——分子生物学家 Agnieszka Obarska-Kosinska 和由生物物理学家 Gerhard Hummer 领导的小组——以及由德国汉堡欧洲分子生物学实验室 (EMBL) 的结构建模师 Jan Kosinski 领导的小组 ,可以更准确地预测人类版本的 Nup 蛋白的形状。 通过利用帮助 AlphaFold 模拟蛋白质相互作用的调整,他们在去年 10 月成功发布了一个模型,涵盖了 60% 的复合体3。 它揭示了复合物如何稳定原子核中的孔,并暗示复合物如何控制进出的东西。
DeepMind’s AI predicts structures for a vast trove of proteins
DeepMind 的 AI 预测大量蛋白质的结构
In the past half-year, AlphaFold mania has gripped the life sciences. “Every meeting I’m in, people are saying ‘why not use AlphaFold?’,” says Christine Orengo, a computational biologist at University College London.
在过去的半年里,AlphaFold 狂热席卷了生命科学领域。 “我参加的每次会议,人们都在说‘为什么不使用 AlphaFold?’,”伦敦大学学院的计算生物学家 Christine Orengo 说。
In some cases, the AI has saved scientists time; in others it has made possible research that was previously inconceivable or wildly impractical. It has limitations, and some scientists are finding its predictions to be too unreliable for their work. But the pace of experimentation is frenetic.
在某些情况下,人工智能为科学家节省了时间; 在其他情况下,它使以前难以想象或非常不切实际的研究成为可能。 它有局限性,一些科学家发现它的预测对于他们的工作来说太不可靠了。 但实验的步伐是狂热的。
Even those who developed the software are struggling to keep up with its use in areas ranging from drug discovery and protein design to the origins of complex life. “I wake up and type AlphaFold into Twitter,” says John Jumper, who leads the AlphaFold team at DeepMind. “It’s quite the experience to see everything.”
即使是开发该软件的人也在努力跟上它在从药物发现和蛋白质设计到复杂生命起源等领域的使用。 “我醒来并在 Twitter 上输入 AlphaFold,”领导 DeepMind AlphaFold 团队的 John Jumper 说。 “看到一切都是一种体验。”
A startling success
惊人的成功
AlphaFold caused a sensation in December 2020, when it dominated a contest called the Critical Assessment of Protein Structure Prediction, or CASP. The competition, held every two years, measures progress in one of biology’s grandest challenges: determining the 3D shapes of proteins from their amino-acid sequence alone. Computer-software entries are judged against structures of the same proteins determined using experimental methods such as X-ray crystallography or cryo-electron microscopy (cryo-EM), which fire X-rays or electron beams at proteins to build up a picture of their shape.
AlphaFold 在 2020 年 12 月引起了轰动,当时它主导了一场名为“蛋白质结构预测关键评估”(CASP)的比赛。 该竞赛每两年举行一次,旨在衡量生物学最大挑战之一的进展:仅从蛋白质的氨基酸序列中确定蛋白质的 3D 形状。 计算机软件条目是根据使用 X 射线晶体学或低温电子显微镜 (cryo-EM) 等实验方法确定的相同蛋白质的结构来判断的,这些方法向蛋白质发射 X 射线或电子束以建立它们的图像 形状。
The 2020 version of AlphaFold was the software’s second edition. It had also won the 2018 CASP, but its earlier efforts mostly weren’t good enough to stand in for experimentally determined structures, says Jumper. However, AlphaFold2’s predictions were, on average, on par with the empirical structures.
AlphaFold 的 2020 版是该软件的第二版。 Jumper 说,它还赢得了 2018 年的 CASP,但其早期的努力大多不足以代替实验确定的结构。 然而,AlphaFold2 的预测平均而言与经验结构相当。
It wasn’t clear when DeepMind would make the software or its predictions widely available, so researchers used information from a public talk by Jumper, and their own insights, to develop their own AI tool, called RoseTTAFold.
目前尚不清楚 DeepMind 何时会广泛使用该软件或其预测,因此研究人员利用 Jumper 公开演讲的信息和他们自己的见解,开发了自己的 AI 工具,称为 RoseTTAFold。
Then on 15 July 2021, papers describing RoseTTAFold and AlphaFold2 appeared2,4, along with freely available, open-source code and other information needed for specialists to run their own versions of the tools. A week later, DeepMind announced that it had used AlphaFold to predict the structure of nearly every protein made by humans, as well as the entire ‘proteomes’ of 20 other widely studied organisms, such as mice and the bacterium Escherichia coli — more than 365,000 structures in total (see ‘What’s known about proteomes’). DeepMind also publicly released these to a database maintained by the EMBL’s European Bioinformatics Institute (EMBL–EBI), in Hinxton, UK. That database has since swelled to almost one million structures.
然后在 2021 年 7 月 15 日,出现了描述 RoseTTAFold 和 AlphaFold2 的论文2、4,以及免费提供的开源代码和专家运行他们自己的工具版本所需的其他信息。 一周后,DeepMind 宣布它已经使用 AlphaFold 预测了人类制造的几乎所有蛋白质的结构,以及其他 20 种广泛研究的生物体的整个“蛋白质组”,例如小鼠和大肠杆菌——超过 365,000 总结构(参见“关于蛋白质组的已知信息”)。 DeepMind 还将这些信息公开发布到由位于英国欣克斯顿的 EMBL 欧洲生物信息学研究所 (EMBL-EBI) 维护的数据库中。 此后,该数据库已膨胀到近一百万个结构。

What’s known about proteomes: bar chart of percentage of structures from different species that come from PDB and AlphaFold.
关于蛋白质组的已知信息:来自 PDB 和 AlphaFold 的不同物种的结构百分比条形图。
Source: E. Porta-Pardo et al. PLoS Comput. Biol. 18, e1009818 (2022).
This year, DeepMind plans to release a total of more than 100 million structure predictions. That is nearly half of all known proteins — and hundreds of times more than the number of experimentally determined proteins in the Protein Data Bank (PDB) structure repository.
今年,DeepMind 计划发布总计超过 1 亿个结构预测。 这几乎是所有已知蛋白质的一半,是蛋白质数据库 (PDB) 结构库中实验确定的蛋白质数量的数百倍。
AlphaFold deploys deep-learning neural networks: computational architectures inspired by the brain’s neural wiring to discern patterns in data. It has been trained on hundreds of thousands of experimentally determined protein structures and sequences in the PDB and other databases. Faced with a new sequence, it first looks for related sequences in databases, which can identify amino acids that have tended to evolve together, suggesting they’re close in 3D space. The structure of existing related proteins provides another way to estimate distances between amino-acid pairs in the new sequence.
AlphaFold 部署了深度学习神经网络:受大脑神经线路启发的计算架构,可识别数据中的模式。 它已经接受了 PDB 和其他数据库中数十万个实验确定的蛋白质结构和序列的训练。 面对一个新序列,它首先在数据库中寻找相关序列,这些序列可以识别出倾向于一起进化的氨基酸,表明它们在 3D 空间中很接近。 现有相关蛋白质的结构提供了另一种估计新序列中氨基酸对之间距离的方法。
AlphaFold iterates clues from these parallel tracks back and forth as it tries to model the 3D positions of amino acids, continually updating its estimate. Specialists say the software’s application of new ideas in machine learning research seems to be what makes AlphaFold so good — in particular, its use of an AI mechanism termed ‘attention’ to determine which amino-acid connections are most salient for its task at any moment.
AlphaFold 在尝试对氨基酸的 3D 位置进行建模时来回迭代来自这些平行轨迹的线索,并不断更新其估计值。 专家表示,该软件在机器学习研究中的新思想应用似乎是 AlphaFold 如此出色的原因——特别是,它使用一种称为“注意力”的人工智能机制来确定哪些氨基酸连接在任何时候对其任务最重要 .
DeepMind’s AI for protein structure is coming to the masses
DeepMind 的蛋白质结构 AI 即将普及
The network’s reliance on information about related protein sequences means that AlphaFold has some limitations. It is not designed to predict the effect of mutations, such as those that cause disease, on a protein’s shape. Nor was it trained to determine how proteins change shape in the presence of other interacting proteins, or molecules such as drugs. But its models come with scores that gauge the network’s confidence in its prediction for each amino-acid unit of a protein — and researchers are tweaking AlphaFold’s code to expand its capabilities.
该网络对相关蛋白质序列信息的依赖意味着 AlphaFold 存在一些局限性。 它并非旨在预测突变(例如导致疾病的突变)对蛋白质形状的影响。 它也没有被训练来确定在其他相互作用的蛋白质或药物等分子存在的情况下蛋白质如何改变形状。 但它的模型附带的分数可以衡量网络对其预测蛋白质每个氨基酸单元的信心——研究人员正在调整 AlphaFold 的代码以扩展其功能。
By now, more than 400,000 people have used the EMBL-EBI’s AlphaFold database, according to DeepMind. There are also AlphaFold ‘power users’: researchers who’ve set up the software on their own servers or turned to cloud-based versions of AlphaFold to predict structures not in the EMBL-EBI database, or to dream up new uses for the tool.
据 DeepMind 称,到目前为止,已有超过 40 万人使用了 EMBL-EBI 的 AlphaFold 数据库。 还有 AlphaFold 的“超级用户”:研究人员在自己的服务器上安装了软件,或者转向基于云的 AlphaFold 版本来预测不在 EMBL-EBI 数据库中的结构,或者为该工具设想新用途 .
Solving structures
求解结构
Biologists are already impressed with AlphaFold’s ability to solve structures. “Based on what I’ve seen so far, I trust AlphaFold quite a lot,” says Thomas Boesen, a structural biologist at Aarhus University in Denmark. The software has successfully predicted the shapes of proteins that Boesen’s centre has determined but not yet published. “That’s a big validation from my side,” he says. He and Aarhus microbial ecologist Tina Šantl-Temkiv are using AlphaFold to model the structure of bacterial proteins that promote the formation of ice — and which could contribute to the cooling effects of ice in clouds — because biologists haven’t been able to fully determine the structures experimentally5.
AlphaFold 解析结构的能力已经给生物学家留下了深刻的印象。 “根据我目前所见,我非常信任 AlphaFold,”丹麦奥胡斯大学的结构生物学家 Thomas Boesen 说。 该软件已成功预测了 Boesen 中心已确定但尚未发表的蛋白质形状。 “这对我来说是一个很大的验证,”他说。 他和奥胡斯微生物生态学家 Tina Šantl-Temkiv 正在使用 AlphaFold 来模拟细菌蛋白质的结构,这些蛋白质促进冰的形成——这可能有助于云中冰的冷却效应——因为生物学家还不能完全确定 结构实验5。
As long as a protein curls up into a single well-defined 3D shape — and not all do — AlphaFold’s prediction can be hard to beat, says Arne Elofsson, a protein bioinformatician at Stockholm University. “It’s a one-click solution to get probably the best model you’re going to get.”
斯德哥尔摩大学的蛋白质生物信息学家 Arne Elofsson 说,只要一种蛋白质卷曲成一个定义明确的 3D 形状——而且并非全部如此——AlphaFold 的预测就很难被击败。 “这是一种一键式解决方案,可能是您将获得的最佳模型。”
Where AlphaFold is less confident, “it’s very good at telling you when it doesn’t work”, Elofsson says. In such cases, predicted structures can resemble floating spaghetti strands (see ‘The good, the bad and the ugly’). This often corresponds to regions of proteins that lack a defined shape, at least in isolation. Such intrinsically disordered regions — which make up around one-third of the human proteome — might become well defined only when another molecule, such as a signalling partner, is present.
Elofsson 说,在 AlphaFold 不太自信的地方,“它非常擅长告诉你什么时候它不起作用”。 在这种情况下,预测的结构可能类似于浮动的意大利面条线(参见“好的、坏的和丑陋的”)。 这通常对应于缺乏确定形状的蛋白质区域,至少在隔离时是这样。 这种本质上无序的区域——约占人类蛋白质组的三分之一——可能只有在存在另一种分子(如信号伙伴)时才能得到很好的定义。

The good, the bad and the ugly: graphic that shows the varying accuracies of AlphaFold’s predictions with confidence estimates.
好的、坏的和丑陋的:图表显示了 AlphaFold 预测的不同准确性和置信度估计。
Images: J. M. Thornton et al. Nature Med. 27, 1666–1669 (2021).
Norman Davey, a computational biologist at the Institute of Cancer Research in London, says AlphaFold’s ability to identify disorder has been a game-changer for his work studying the properties of these regions. “Instantly there was a huge increase in the quality of the predictions we had, without any effort on our part,” he says.
伦敦癌症研究所的计算生物学家 Norman Davey 表示,AlphaFold 识别疾病的能力已经改变了他研究这些区域特性的工作。 他说:“我们的预测质量立即有了巨大的提高,而我们没有付出任何努力。”
AlphaFold’s dump of protein structures into the EMBL-EBI database is also immediately being put to use. Orengo’s team is searching it to identify fresh kinds of proteins (without experimentally verifying them) and has turned up hundreds, perhaps thousands, of potentially new protein families, expanding scientists’ knowledge of what proteins look like and can do. In another effort, the team is scouring databases of DNA sequences harvested from the ocean and waste water, to try to identify new plastic-eating enzymes. Using AlphaFold to quickly approximate the structures of thousands of proteins, the researchers hope to better understand how enzymes evolved to break down plastic, and potentially to improve them.
AlphaFold 将蛋白质结构转储到 EMBL-EBI 数据库中的数据也立即投入使用。 Orengo 的团队正在搜索它以识别新的蛋白质种类(没有通过实验验证它们),并且已经发现了数百甚至数千个潜在的新蛋白质家族,扩大了科学家对蛋白质外观和功能的了解。 在另一项努力中,该团队正在搜索从海洋和废水中采集的 DNA 序列数据库,以尝试识别新的食用塑料酶。 使用 AlphaFold 快速近似数千种蛋白质的结构,研究人员希望更好地了解酶如何进化以分解塑料,并有可能改进它们。
The ability to transform any protein-coding gene sequence into a reliable structure should be especially powerful for evolution studies, says Sergey Ovchinnikov, an evolutionary biologist at Harvard University in Cambridge, Massachusetts. Researchers compare genetic sequences to determine how organisms and their genes are related across species. For distantly related genes, comparisons might fail to turn up evolutionary relatives because the sequences have changed so much. But by comparing protein structures — which tend to change less rapidly than genetic sequences — researchers might be able to uncover overlooked ancient relationships. “This opens up an amazing opportunity to study the evolution of proteins and the origins of life,” says Pedro Beltrao, a computational biologist at the Swiss Federal Institute of Technology in Zurich.
马萨诸塞州剑桥市哈佛大学的进化生物学家 Sergey Ovchinnikov 说,将任何蛋白质编码基因序列转化为可靠结构的能力对于进化研究来说应该是特别强大的。 研究人员比较基因序列以确定生物及其基因在物种间的相关性。 对于远缘相关的基因,比较可能无法找到进化亲属,因为序列发生了很大变化。 但通过比较蛋白质结构——其变化往往不如基因序列快——研究人员或许能够发现被忽视的古老关系。 苏黎世瑞士联邦理工学院的计算生物学家 Pedro Beltrao 说:“这为研究蛋白质进化和生命起源提供了一个绝佳的机会。”
To test this idea, a team led by Martin Steinegger, a computational biologist at Seoul National University, and his colleagues used a tool they developed, called Foldseek, to look for relatives of the RNA-copying enzyme of SARS-CoV-2 — the virus that causes COVID-19 — in the EMBL-EBI’s AlphaFold database6. This search turned up previously unidentified possible ancient relatives: proteins across eukaryotes — including slime moulds — that resemble, in their 3D structure, enzymes called reverse transcriptases that viruses such as HIV use to copy RNA into DNA, despite very little similarity at the genetic-sequence level.
为了验证这个想法,首尔国立大学的计算生物学家 Martin Steinegger 和他的同事领导的一个团队使用他们开发的名为 Foldseek 的工具来寻找 SARS-CoV-2 的 RNA 复制酶的亲属—— 导致 COVID-19 的病毒——在 EMBL-EBI 的 AlphaFold 数据库中6。 这项搜索发现了以前未知的可能的远古亲属:真核生物中的蛋白质——包括粘菌——在它们的 3D 结构中类似于称为逆转录酶的酶,病毒如 HIV 使用逆转录酶将 RNA 复制到 DNA 中,尽管在遗传上几乎没有相似性。 序列级别。
Experimental assistant
For scientists who want to determine the detailed structure of a specific protein, an AlphaFold prediction isn’t necessarily an immediate solution. Rather, it provides an initial approximation that can be validated or refined by experiment — and which itself helps to make sense of experimental data. Raw data from X-ray crystallography, for instance, appear as patterns of diffracted X-rays. Typically, scientists need a starting guess at a protein’s structure to interpret these patterns. Previously, they’d often cobble together information from related proteins in the PDB or use experimental approaches, says Randy Read, a structural biologist at the University of Cambridge, UK, whose lab specialized in some of these methods. Now, AlphaFold’s predictions have rendered such approaches unnecessary for most X-ray patterns, Read says, and his lab is working to make better use of AlphaFold in experimental models. “We’ve totally refocused our research.”
对于想要确定特定蛋白质的详细结构的科学家来说,AlphaFold 预测不一定是立竿见影的解决方案。 相反,它提供了一个可以通过实验验证或改进的初始近似值——它本身有助于理解实验数据。 例如,来自 X 射线晶体学的原始数据显示为衍射 X 射线的图案。 通常,科学家需要对蛋白质结构进行初步猜测才能解释这些模式。 英国剑桥大学的结构生物学家 Randy Read 说,以前,他们经常将 PDB 中相关蛋白质的信息拼凑在一起,或者使用实验方法。他的实验室专门研究其中一些方法。 现在,AlphaFold 的预测使得大多数 X 射线模式不需要这种方法,Read 说,他的实验室正在努力在实验模型中更好地利用 AlphaFold。 “我们完全重新调整了研究重点。”
Artificial intelligence powers protein-folding predictions
人工智能为蛋白质折叠预测提供动力
He and other researchers have used AlphaFold to determine crystal structures from X-ray data that were uninterpretable without an adequate starting model. “People are solving structures that, for years, had not been solved,” says Claudia Millán Nebot, a former postdoc in Read’s lab who now works at the analytics firm SciBite in Cambridge. She expects to see a glut of new protein structures submitted to the PDB, in large part as a result of AlphaFold.
他和其他研究人员已经使用 AlphaFold 从 X 射线数据中确定晶体结构,这些数据在没有足够的起始模型的情况下是无法解释的。 “人们正在解决多年来一直没有解决的结构,”Claudia Millán Nebot 说,他是 Read 实验室的前博士后,现在在剑桥的分析公司 SciBite 工作。 她预计会看到大量新的蛋白质结构提交给 PDB,这在很大程度上是 AlphaFold 的结果。
The same is true for labs specializing in cryo-EM, which captures pictures of flash-frozen proteins. In some instances, AlphaFold’s models have accurately predicted unique features of proteins called G-protein-coupled receptors (GPCRs) — which are important drug targets — that other computational tools got wrong, says Bryan Roth, a structural biologist and pharmacologist at the University of North Carolina at Chapel Hill. “It seems to be really good for generating first models, which we then refine with some experimental data,” he says. “That saves us some time.”
专门从事冷冻电镜研究的实验室也是如此,它可以捕捉快速冷冻蛋白质的照片。 在某些情况下,AlphaFold 的模型准确地预测了称为 G 蛋白偶联受体 (GPCR) 的蛋白质的独特特征,这些蛋白质是重要的药物靶标,而其他计算工具出错了,美国大学的结构生物学家和药理学家 Bryan Roth 说 北卡罗来纳州教堂山。 “它似乎非常适合生成第一个模型,然后我们用一些实验数据对其进行改进,”他说。 “这为我们节省了一些时间。”
But Roth adds that AlphaFold isn’t always that accurate. Of the several dozen GPCR structures his lab has solved, but not yet published, he says, “about half the time, the AlphaFold structures are fairly good, and half the time they’re more or less useless for our purposes”. In some instances, he says, AlphaFold labels predictions with high confidence, but experimental structures show that it is wrong. Even when the software gets it right, it cannot model how a protein would look when bound to a drug or other small molecule (ligand), which can substantially alter the structure. Such caveats make Roth wonder how useful AlphaFold will be for drug discovery.
但 Roth 补充说,AlphaFold 并不总是那么准确。 他说,在他的实验室已经解决但尚未发表的几十个 GPCR 结构中,“大约有一半的时间,AlphaFold 结构相当好,而有一半的时间它们或多或少对我们的目的毫无用处”。 他说,在某些情况下,AlphaFold 以高置信度标记预测,但实验结构表明它是错误的。 即使软件做对了,它也无法模拟蛋白质与药物或其他小分子(配体)结合时的外观,这会大大改变结构。 这些警告让 Roth 想知道 AlphaFold 对药物发现有多大用处。
It’s increasingly common in drug-discovery efforts to use computational-docking software that screens billions of small molecules to find some that might bind to proteins — one indication that they could make useful drugs. Roth is now working with Brian Shoichet, a medicinal chemist at the University of California, San Francisco, to see how AlphaFold’s predictions compare with experimentally determined structures in this exercise.
在药物发现工作中,使用计算对接软件越来越普遍,该软件可以筛选数十亿个小分子,以找到一些可能与蛋白质结合的分子——这表明它们可以制造有用的药物。 Roth 现在正与加州大学旧金山分校的药物化学家 Brian Shoichet 合作,以了解 AlphaFold 的预测如何与本练习中通过实验确定的结构进行比较。
Shoichet says they are limiting their work to proteins for which AlphaFold’s prediction chimes with experimental structures. But even in these instances, the docking software is turning up different drug hits for the experimental structure and AlphaFold’s take, suggesting that small discrepancies could matter. “That doesn’t mean we won’t find new ligands, we’ll just find different ones,” says Shoichet. His team is now synthesizing potential drugs identified using AlphaFold structures, and testing their activity in the lab.
Shoichet 说,他们将工作限制在 AlphaFold 的预测与实验结构相吻合的蛋白质上。 但即使在这些情况下,对接软件也会为实验结构和 AlphaFold 提供不同的药物命中率,这表明微小的差异可能很重要。 “这并不意味着我们不会找到新的配体,我们只会找到不同的配体,”Shoichet 说。 他的团队现在正在合成使用 AlphaFold 结构识别的潜在药物,并在实验室中测试它们的活性。
Critical optimism
Researchers at pharmaceutical companies and biotechnology firms are excited about AlphaFold’s potential to help with drug discovery, says Shoichet. “Critical optimism is how I’d describe it.” In November 2021, DeepMind launched its own spin-off, Isomorphic Labs, which aims to apply AlphaFold and other AI tools to drug discovery. But the company has said little else about its plans.
Shoichet 说,制药公司和生物技术公司的研究人员对 AlphaFold 帮助药物发现的潜力感到兴奋。 “批判性的乐观是我描述它的方式。” 2021 年 11 月,DeepMind 推出了自己的衍生产品 Isomorphic Labs,旨在将 AlphaFold 和其他 AI 工具应用于药物发现。 但该公司对其计划只字未提。
Karen Akinsanya, who leads therapeutics development at Schrödinger, a drug-discovery firm headquartered in New York City that also publishes chemical-simulations software, says she and her colleagues are already having some success using AlphaFold structures, including for GPCRs, in virtual screens and compound design for drug candidates. She finds that, just as with experimental structures, extra software is needed to get at the fine details of amino-acid side chains or locations where individual hydrogen atoms might sit. Once this is done, AlphaFold structures have proved good enough to guide drug discovery — in some cases.
Karen Akinsanya 是 Schrödinger 的治疗开发负责人,Schrödinger 是一家总部位于纽约市的药物发现公司,还发布了化学模拟软件,她说她和她的同事已经在虚拟屏幕和 GPCR 中使用 AlphaFold 结构取得了一些成功。 候选药物的化合物设计。 她发现,就像实验结构一样,需要额外的软件来获取氨基酸侧链或单个氢原子可能所在位置的详细信息。 一旦完成,AlphaFold 结构已被证明足以指导药物发现——在某些情况下。
“It’s hard to say ‘this is a panacea’; that because you can do it very well for one structure — surprisingly and excitingly well — that it is eminently applicable to all structures. It clearly isn’t,” Akinsanya says. And she and her colleagues have found that AlphaFold’s accuracy predictions don’t show whether a structure will be useful for later drug screening. AlphaFold structures will never fully replace experimental ones in drug discovery, she says. But they might speed up the process by complementing experimental methods.
“很难说‘这是灵丹妙药’; 因为你可以为一个结构做得很好——令人惊讶和令人兴奋的——它非常适用于所有结构。 显然不是,”Akinsanya 说。 她和她的同事发现,AlphaFold 的准确性预测并不能显示一个结构是否对以后的药物筛选有用。 她说,AlphaFold 结构永远不会完全取代药物发现中的实验性结构。 但他们可能会通过补充实验方法来加速这一过程。
Drug developers curious about AlphaFold received good news in January, when DeepMind lifted a key restriction on its use for commercial applications. When the company released AlphaFold’s code in July 2021, it had stipulated that the parameters, or weights, needed to run the AlphaFold neural network — the end result of training the network on hundreds of thousands of protein structures and sequences — were for non-commercial use only. Akinsanya says this was a bottleneck for some in industry, and there was a “wave of excitement” when DeepMind changed tack. (RoseTTAFold came with similar restrictions, says Ovchinnikov, one of its developers. But the next version will be fully open-source.)
对 AlphaFold 感到好奇的药物开发人员在 1 月份收到了好消息,当时 DeepMind 取消了对其用于商业应用的关键限制。 当该公司在 2021 年 7 月发布 AlphaFold 的代码时,它规定运行 AlphaFold 神经网络所需的参数或权重——这是在数十万个蛋白质结构和序列上训练网络的最终结果——用于非商业用途 仅使用。 Akinsanya 说,这对行业中的一些人来说是一个瓶颈,当 DeepMind 改变策略时出现了一股“兴奋的浪潮”。 (RoseTTAFold 也有类似的限制,其开发人员之一 Ovchinnikov 说。但下一个版本将完全开源。)
AI tools are not just changing how scientists determine what proteins look like. Some researchers are using them to make entirely new proteins. “Deep learning is completely transforming the way that protein design is being done in my group,” says David Baker, a biochemist at the University of Washington in Seattle and a leader in the field of designing proteins, as well as predicting their structures. His team, with computational chemist Minkyung Baek, led the work to develop RoseTTAFold.
人工智能工具不仅改变了科学家确定蛋白质外观的方式。 一些研究人员正在使用它们来制造全新的蛋白质。 “深度学习正在彻底改变我团队中蛋白质设计的方式,”西雅图华盛顿大学的生物化学家、蛋白质设计和预测其结构领域的领导者大卫贝克说。 他的团队与计算化学家 Minkyung Baek 一起领导了开发 RoseTTAFold 的工作。
Baker’s team gets AlphaFold and RoseTTAFold to “hallucinate” new proteins. The researchers have altered the AI code so that, given random sequences of amino acids, the software will optimize them until they resemble something that the neural networks recognize as a protein (see ‘Dreaming up proteins’).
Baker 的团队让 AlphaFold 和 RoseTTAFold 能够“产生幻觉”新的蛋白质。 研究人员已经改变了人工智能代码,因此,给定氨基酸的随机序列,软件将对其进行优化,直到它们类似于神经网络识别为蛋白质的东西(参见“梦想蛋白质”)。
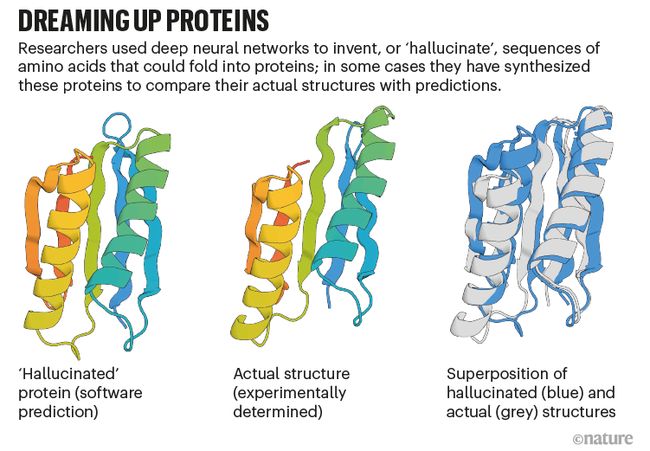
Dreaming up proteins: graphic that compares a protein structure predicted by a neural network with an actual stucture.
梦想蛋白质:将神经网络预测的蛋白质结构与实际结构进行比较的图形。
Images: Ref. 7
In December 2021, Baker and his colleagues reported expressing 129 of these hallucinated proteins in bacteria, and found that about one-fifth of them folded into something resembling their predicted shape7. “That’s really the first demonstration that you can design proteins using these networks,” Baker says. His team is now using this approach to design proteins that do useful things, such as catalyse a particular chemical reaction, by specifying the amino acids responsible for the desired function and letting the AI dream up the rest.
2021 年 12 月,贝克和他的同事报告说,在细菌中表达了 129 种这些幻觉蛋白,并发现其中约五分之一折叠成类似于其预测形状的东西 7。 “这确实是第一次证明你可以使用这些网络设计蛋白质,”贝克说。 他的团队现在正在使用这种方法来设计做有用事情的蛋白质,例如催化特定的化学反应,方法是指定负责所需功能的氨基酸,并让 AI 梦想其余部分。
Animation of four protein structures being predicted by the Alphafold AI system
Alphafold AI 系统预测的四种蛋白质结构的动画
Four examples of protein ‘hallucination’. In each case, AlphaFold is presented with a random amino-acid sequence, predicts the structure, and changes the sequence until the software confidently predicts that it will fold into a protein with a well-defined 3D shape. Colours show prediction confidence (from red for very low confidence, through yellow and light blue to dark blue for very high confidence). Initial frames have been slowed down for clarity.Credit: Sergey Ovchinnikov
蛋白质“幻觉”的四个例子。 在每种情况下,AlphaFold 都会显示一个随机氨基酸序列,预测结构并更改序列,直到软件有把握地预测它将折叠成具有明确 3D 形状的蛋白质。 颜色显示预测置信度(从红色表示非常低的置信度,通过黄色和浅蓝色到深蓝色表示非常高的置信度)。 为了清晰起见,最初的帧已经放慢了。Credit: Sergey Ovchinnikov
Hacking AlphaFold
When DeepMind released its AlphaFold code, Ovchinnikov wanted to better understand how the tool worked. Within days, he and computational-biology colleagues, including Steinegger, set up a website called ColabFold that allowed anyone to submit a protein sequence to AlphaFold or RoseTTAFold and get a structure prediction. Ovchinnikov imagined that he and other scientists would use ColabFold to try and ‘break’ AlphaFold, for instance, by supplying false information about a target protein sequence’s evolutionary relatives. By doing this, Ovchinnikov hoped he could determine how the network had learnt to predict structures so well.
当 DeepMind 发布其 AlphaFold 代码时,Ovchinnikov 想要更好地了解该工具的工作原理。 几天之内,他和包括 Steinegger 在内的计算生物学同事建立了一个名为 ColabFold 的网站,允许任何人向 AlphaFold 或 RoseTTAFold 提交蛋白质序列并获得结构预测。 Ovchinnikov 设想他和其他科学家会使用 ColabFold 来尝试“破坏”AlphaFold,例如,通过提供有关目标蛋白质序列进化亲属的虚假信息。 通过这样做,Ovchinnikov 希望他能够确定网络是如何学会如此出色地预测结构的。
As it turned out, most researchers who used ColabFold just wanted to get a protein structure. But others used it as a platform to modify the inputs to AlphaFold to tackle new applications. “I didn’t expect the number of hacks of various types,” says Jumper.
事实证明,大多数使用 ColabFold 的研究人员只是想获得蛋白质结构。 但其他人将其用作修改 AlphaFold 的输入以处理新应用程序的平台。 “我没想到会出现各种类型的黑客攻击,”Jumper 说。
By far the most popular hack has been to wield the tool on protein complexes comprised of multiple, interacting — and often intertwined — chains of peptides. Just as with the nuclear pore complex, many proteins in cells gain their function when they form complexes with multiple protein subunits.
到目前为止,最流行的黑客攻击是在蛋白质复合物上使用该工具,该复合物由多个相互作用的——通常是相互交织的——肽链组成。 就像核孔复合物一样,细胞中的许多蛋白质在与多个蛋白质亚基形成复合物时发挥作用。
AlphaFold was designed to predict the shape of single peptide chains, and its training consisted entirely of such proteins. But the network seems to have learnt something about how complexes fold together. Several days after AlphaFold’s code was released, Yoshitaka Moriwaki, a protein bioinformatician at the University of Tokyo, tweeted that it could accurately predict interactions between two protein sequences if they were stitched together with a long linker sequence. Baek soon shared another hack to predict complexes, gleaned from developing RoseTTAFold.
AlphaFold 旨在预测单个肽链的形状,其训练完全由此类蛋白质组成。 但该网络似乎已经了解了一些关于复合物如何折叠在一起的知识。 AlphaFold 的代码发布几天后,东京大学的蛋白质生物信息学家 Yoshitaka Moriwaki 在推特上表示,如果将两个蛋白质序列与长连接序列缝合在一起,它可以准确预测它们之间的相互作用。 Baek 很快分享了另一个从开发 RoseTTAFold 中收集到的预测复合物的技巧。
ColabFold later incorporated the ability to predict complexes. And in October 2021, DeepMind released an update called AlphaFold-Multimer8 that was specifically trained on protein complexes, unlike its predecessor. Jumper’s team applied it to thousands of complexes in the PDB, and found that it predicted around 70% of the known protein–protein interactions.
ColabFold 后来加入了预测复合物的能力。 并且在 2021 年 10 月,DeepMind 发布了一个名为 AlphaFold-Multimer8 的更新,该更新专门针对蛋白质复合物进行训练,与其前身不同。 Jumper 的团队将其应用于 PDB 中的数千个复合物,发现它预测了大约 70% 的已知蛋白质-蛋白质相互作用。
These tools are already helping researchers to spot potential new protein partners. Elofsson’s team used AlphaFold to predict the structures of 65,000 human protein pairs that were suspected to interact on the basis of experimental data9. And a team led by Baker used AlphaFold and RoseTTAFold to model interactions between nearly every pair of proteins encoded by yeast, identifying more than 100 previously unknown complexes10. Such screens are just starting points, says Elofsson. They do a good job of predicting some protein pairings, particularly those that are stable, but struggle to identify more transient interactions. “Because it looks nice doesn’t mean it is correct,” says Elofsson. “You need some experimental data that show you’re right.”
这些工具已经在帮助研究人员发现潜在的新蛋白质伙伴。 Elofsson 的团队使用 AlphaFold 预测了 65,000 个人类蛋白质对的结构,这些蛋白质对根据实验数据被怀疑相互作用9。 Baker 领导的一个团队使用 AlphaFold 和 RoseTTAFold 来模拟酵母编码的几乎每一对蛋白质之间的相互作用,识别出 100 多个以前未知的复合物 10。 Elofsson 说,这样的屏幕只是起点。 他们在预测某些蛋白质配对方面做得很好,尤其是那些稳定但难以识别更多瞬时相互作用的蛋白质配对。 “因为它看起来不错并不意味着它是正确的,”Elofsson 说。 “你需要一些实验数据来证明你是对的。”
The nuclear pore complex work is a good example of how predictions and experimental data can work together, says Kosinski (see ‘Genome gateway’). “It’s not like we take all the 30 proteins, throw them into AlphaFold and get the structure out.” To put the predicted protein structures together, the team used 3D images of the nuclear pore complex, captured using a form of cryo-EM called cryo-electron tomography. In one instance, experiments that can determine the proximity of proteins turned up a surprising interaction between two components of the complex, which AlphaFold’s models then confirmed.
Kosinski 说,核孔复合体工作是预测和实验数据如何协同工作的一个很好的例子(参见“基因组网关”)。 “这并不是说我们将所有 30 种蛋白质都放入 AlphaFold 中并取出结构。” 为了将预测的蛋白质结构组合在一起,该团队使用了核孔复合物的 3D 图像,这些图像是使用一种称为低温电子断层扫描的低温电子显微镜拍摄的。 在一个例子中,可以确定蛋白质接近度的实验在复合物的两个成分之间产生了令人惊讶的相互作用,AlphaFold 的模型随后证实了这一点。

Genome gateway: Two views of the human nuclear pore complex showing how it embeds in the nuclear membrane
基因组网关:人类核孔复合体的两种视图显示它如何嵌入核膜
Images adapted from ref. 3/Agnieszka Obarska-Kosinska
Kosinski sees the team’s current map of the nuclear pore complex as a starting point for experiments and simulations that examine how the pore complex functions — and how it malfunctions in disease.
Kosinski 将团队当前的核孔复合体地图视为实验和模拟的起点,这些实验和模拟检查了孔复合体的功能 - 以及它如何在疾病中出现故障。
AlphaFold’s limits
For all the progress made with AlphaFold, scientists say that it is important to be clear about its limitations — particularly because researchers who don’t specialize in predicting protein structures use it.
对于 AlphaFold 取得的所有进展,科学家们表示,重要的是要清楚它的局限性——特别是因为不专门预测蛋白质结构的研究人员会使用它。
Attempts to apply AlphaFold to various mutations that disrupt a protein’s natural structure, including one linked to early breast cancer, have confirmed that the software is not equipped to predict the consequences of new mutations in proteins, since there are no evolutionarily-related sequences to examine11.
尝试将 AlphaFold 应用于破坏蛋白质自然结构的各种突变,包括与早期乳腺癌相关的突变,已证实该软件无法预测蛋白质新突变的后果,因为没有进化相关的序列可供检查11 .
The AlphaFold team is now thinking about how a neural network could be designed to deal with new mutations. Jumper expects this would require the network to better predict how a protein goes from its unfolded to its folded state. That would probably need software that relies only on what it has learnt about protein physics to predict structures, says Mohammed AlQuraishi, a computational biologist at Columbia University in New York City. “One thing we are interested in is making predictions from single sequences without using evolutionary information,” he says. “That’s a key problem that does remain open.”
AlphaFold 团队现在正在考虑如何设计神经网络来处理新的突变。 Jumper 预计这将需要网络更好地预测蛋白质如何从展开状态变为折叠状态。 纽约市哥伦比亚大学的计算生物学家 Mohammed AlQuraishi 说,这可能需要仅依靠它所学到的蛋白质物理学知识来预测结构的软件。 “我们感兴趣的一件事是在不使用进化信息的情况下从单个序列进行预测,”他说。 “这是一个尚未解决的关键问题。”
AlphaFold is also designed to predict a single structure, although it has been hacked to spit out more than one. But many proteins take on multiple conformations, which can be important to their function. “AlphaFold can’t really deal with proteins that can adopt different structures in different conformations,” says Schueler-Furman. And the predictions are for structures in isolation, whereas many proteins function alongside ligands such as DNA and RNA, fat molecules and minerals such as iron. “We are still missing ligands, we are missing everything else about proteins,” says Elofsson.
AlphaFold 也被设计用来预测一个单一的结构,尽管它已经被黑客破解了不止一个。 但是许多蛋白质具有多种构象,这对其功能可能很重要。 “AlphaFold 不能真正处理可以采用不同构象的不同结构的蛋白质,”Schueler-Furman 说。 并且预测是针对孤立结构的,而许多蛋白质与配体(如 DNA 和 RNA)、脂肪分子和矿物质(如铁)一起发挥作用。 “我们仍然缺少配体,我们缺少关于蛋白质的其他一切,”Elofsson 说。
Developing these next-generation neural networks will be a huge challenge, says AlQuraishi. AlphaFold relied on decades of research which generated experimental structures of proteins that the network could learn from. That volume of data is currently not available to capture protein dynamics, or the shapes of the trillions of smaller molecules that proteins could interact with. The PDB includes structures of proteins as they interact with other molecules, but this captures just a sliver of chemical diversity, Jumper adds.
AlQuraishi 说,开发这些下一代神经网络将是一个巨大的挑战。 AlphaFold 依赖于数十年的研究,这些研究产生了网络可以学习的蛋白质实验结构。 目前无法获得如此大量的数据来捕捉蛋白质动力学,或者蛋白质可以与之相互作用的数万亿个小分子的形状。 Jumper 补充说,PDB 包括蛋白质与其他分子相互作用时的结构,但这仅捕获了一小部分化学多样性。
Researchers think that it will take time for them to determine how best to wield AlphaFold and related AI tools. AlQuraishi sees parallels with the early days of television, when some programmes consisted of radio broadcasters simply reading the news. “I think we’re going to find new applications of structure that we haven’t conceived of yet.”
研究人员认为,他们需要时间来确定如何最好地使用 AlphaFold 和相关的人工智能工具。 AlQuraishi 看到了电视早期的相似之处,当时一些节目由广播电台组成,只是阅读新闻。 “我认为我们将找到我们尚未想到的结构的新应用。”
Where the AlphaFold revolution is ends up is anybody’s guess. “Things are just changing so fast,” says Baker. “Even in the next year, we’re going to see really major breakthroughs made using these tools.” Janet Thornton, a computational biologist at the EMBL-EBI, thinks one of AlphaFold’s biggest impacts might be simply to convince biologists to be more open to insights from computational and theoretical approaches. “To me, the revolution is the mindset change,” she says.
AlphaFold 革命的终点在哪里,谁也说不准。 “事情变化太快了,”贝克说。 “即使在明年,我们也将看到使用这些工具取得的重大突破。” EMBL-EBI 的计算生物学家 Janet Thornton 认为,AlphaFold 的最大影响之一可能只是说服生物学家对计算和理论方法的见解更加开放。 “对我来说,革命就是思维方式的改变,”她说。
The AlphaFold revolution has inspired Kosinski to dream big. He imagines that AlphaFold-inspired tools could be used to model not just individual proteins and complexes, but entire organelles or even cells down to the level of individual protein molecules. “This is the dream we will follow for the next decades.”
AlphaFold 革命激发了 Kosinski 的远大梦想。 他认为受 AlphaFold 启发的工具不仅可用于对单个蛋白质和复合物进行建模,还可以对整个细胞器甚至细胞进行建模,直至单个蛋白质分子的水平。 “这是我们未来几十年的梦想。”
Nature 604, 234-238 (2022)
doi: https://doi.org/10.1038/d41586-022-00997-5
UPDATES & CORRECTIONS
Correction 25 April 2022: An earlier version of this story erroneously described Gerhard Hummer as a biochemist.
2022 年 4 月 25 日更正:这个故事的早期版本错误地将 Gerhard Hummer 描述为生物化学家。
References
- Kosinski, J. et al. Science 5, 363–365 (2016).
- Jumper, J. et al. Nature 596, 583–589 (2021).
- Mosalaganti, S. et al. Preprint at bioRxiv https://doi.org/10.1101/2021.10.26.465776 (2021).
- Hartmann, S. et al. Preprint at bioRxiv https://doi.org/10.1101/2022.01.21.477219 (2022).
- van Kempen, M. et al. Preprint at bioRxiv https://doi.org/10.1101/2022.02.07.479398 (2022).
- Anishchenko, I. et al. Nature 600, 547–552 (2021).
- Evans, R. et al. Preprint at bioRxiv https://doi.org/10.1101/2021.10.04.463034 (2021).
- Bryant, P., Pozzati, G. & Elofsson, A. Nature Commun. 13, 1265 (2022).
- Humphreys, I. R. et al. Science 374, eabm4805 (2021).
- Buel, G. R. & Walters, K. J. Nature Struct. Mol. Biol. 29, 1–2 (2022).