RNAseq生信分析流程简介
文章目录
-
-
- RNAseq简介
- RNAseq分析流程
-
- 1. 实验设计
-
- 1.1 目标RNA提取策略和文库类型选择
- 1.2 测序深度或文库大小
- 1.3 实验重复
- 2. RNAseq信息分析
-
- 2.1 质控 和 过滤
- 2.2 比对
- 2.3 新转录本的发现
- 2.4 基因和转录水平的定量
- 2.5 差异基因表达分析
- 2.6 可变剪接分析
- 2.7 基因融合
-
RNAseq简介
转录组是连接遗传信息与生物功能的桥梁,在广义上指在相同生理条件下的一个或一群细胞中所能转录出的所有RNA的总和,包括编码RNA及非编码RNA;狭义上指所有mRNA的集合[1]。转录组测序分析(RNA-seq)通过提取所要研究的mRNA,将其反转录成cDNA文库,在DNA小片段两端加上接头,利用高通量测序技术统计相关小片段数计算出不同mRNA的表达量,精确地识别可变剪切位点及编码序列单核苷酸多态性,获得某一物种特定组织或器官在某一状态下几乎所有转录本的序列信息[2].目前RNAseq已广泛应用于基础研究、临床诊断和药物研发等领域。
RNAseq分析流程
一般的来讲,RNA-seq的工作流程包含了实验设计、质量控制、比对、基因和转录水平的定量、表达可视化、差异基因筛选、选择性剪接、功能分析、基因融合检测等分析模块[3]。需要注意的是,RNA 测序 (RNA-seq) 具有广泛的应用,目前并没有一种分析流程可以适用于所有情况. 每个 RNA-seq 实验场景都可能有不同的转录本量化、标准化和最终差异表达分析的最佳方法。
1. 实验设计
RNAseq实验设计一般需要考虑三个因素:1)目标RNA提取策略和文库类型选择 2)测序深度或文库大小 3)实验重复。
1.1 目标RNA提取策略和文库类型选择
RNAseq实验的第一步便是提取RNA,由于rRNA 通常占细胞总 RNA 的 90% 以上,而我们感兴趣的信使 RNA (mRNA)往往仅仅只占 1-2%,因此实验中的一个重要步骤便是如何从海量的高丰度核糖体 RNA (rRNA)中提取我们感兴趣的mRNA。目前主要有2种提取方案:1) Poly(A)钓取法和 2) 去rRNA法。Poly(A)钓取法是指通过带有oligo(dT)磁珠与 mRNA的polyA尾巴进行结合,从而将带有polyA尾巴的mRNA提取出来。这种方法往往需要相对较高比例的 mRNA,且降解最小(RNA的降解程度可以通过 RNA 完整值 (RIN) 测量)。然而,许多生物样本(如组织活检)无法获得足够数量或足够好的 mRNA 完整性,因此无法获取到良好的 poly(A) RNA-seq 文库,这种情况下,就需要使用去rRNA法,顾名思义,就是用特定的酶将rRNA裂解掉。
文库选择上可以考虑是否保留链特异性。所谓的链特异性文库,即我们构建出来的cDNA文库保持和原来提取RNA相同的链信息。例如某一条RNA序列来自正义链,序列为“AAATTC”, 构建链特异性的cDNA文库中对于该序列将只会包含“AAATTC”,而不会有其反向互补序列“GAATTT”生成。而非链特异性的文库则两种序列都会存在。这种链特异性的文库通常通过 dUTP 方法实现,通过在第二个 cDNA 合成步骤中加入 UTP 核苷酸来扩增形成互补序列,然后在接头连接之前消化含有 dUTP 的链,达到去除互补链的目的。
文库类型上可以选择单端(SE)或者双端测序(PE)。单端测序通常测得更短,成本更便宜,这种测序类型足以用来研究基因组注释良好的物种的基因表达水平,因此对于研究比较多的模式生物,如果你只想做已知基因的表达量分析,SE是足够了的。但如果你还想研究新的转录本,可变剪切等其他分析时,双端测序往往更好的选择,因为他更长,提供的转录组信息更多。
1.2 测序深度或文库大小
测序深度或文库大小,即给定样本的测序数据量。我们都知道随着样本测序深度增加,检测到的转录本将会更多(更多地表达量的转录本被测到),并且它们的定量结果将会更加精确。具体测多少依然是取决于实验的目的。例如在研究样本复杂性有限单细胞分析中,通常只需1M的read即可进行表达定量,如果你只关注高表达量的基因,甚至20-50K的read数即可很好的定量。在bulk细胞测序中,通常需要5M以上的reads才能准确量化大多数真核转录组中中等到高表达的基因。 如果想精确量化具有低表达水平的基因和转录本,测序量可能要达到100M。 当然这和物种也是有关系的,往往需要具体问题具体分析。我们可以使用测序饱和曲线来评估在给定测序深度下预期的转录组覆盖度分布情况,当曲线逐渐缓和时,表明测序基本达到饱和。
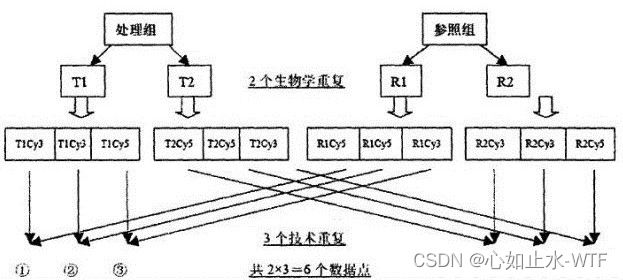
1.3 实验重复
重复可以很好消除偶发误差带来结果偏差,一个好的实验设计通常包含生物学重复和技术重复(两者区别如下图示)
RNA-seq 实验中应包含的重复次数取决于 RNA-seq 中的技术变异量和所研究系统的生物学变异性,以及所需的统计功效(即,检测实验组之间基因表达的统计学显着差异的能力)
2. RNAseq信息分析
RNA-seq 主要分析步骤,包括质控、比对和基因和转录水平的定量、新转录本的发现,差异基因表达,可变剪接分析等等,每个分析都会有非常多可选的软件供选择。下面逐一简要介绍。
2.1 质控 和 过滤
RNAseq的质控往往包含很多方面,测序质控,比对率,测序饱和度,gene覆盖度均一性等等。测序质控是最常见的,其主要是对原始下机数据的质量控制,包含序列质量、GC 含量、接头的存在、过度表达的kmer和重复序列的分析。关于质控的详细信息可以参考 https://rtsf.natsci.msu.edu/genomics/tech-notes/fastqc-tutorial-and-faq/, 对每部分都有基本介绍。主要使用的软件包括:FastQC(对 Illumina 读数执行质控分析的流行工具)和 NGSQC(可以适用于任何平台)。 通过测序质控软件可以了解样本测序质量情况,针对质控的结果考虑是否弃用样本,以及可以针对特定的问题例如接头,N碱基比例高,低质量碱基等进行过滤处理。常用软件保持FASTX-Toolkit,fastp, Cutadapt 和 Trimmomatic 等。 其他质控项比对率,测序饱和度,gene覆盖度均一性等往往需要根据实际经验情况来判断。例如,人类的RNAseq测序数据往往70% 到 90% 数据都会比对到人类基因组上,当然这也取决于所使用的比对工具。当这些质控信息出现异常时需要根据具体的实验等信息分析原因,才能确定解决方案。
2.2 比对
当物种已存在可用的参考序列时,有两种比对策略可供选择:1)直接将测序read 比对到参考基因组 2)比对到参考转录组上。比对到参考基因组时,由于有转录后剪切(可变剪切)的存在,在序列比对时需要充分考虑gap。常用的软件为 TopHat2,HISAT, STAR等。当我们不需要发现新的转录本时,我们可以将测序数据直接比对到参考转录组上。
当研究物种没有可用的基因组时,首先需要将 reads 组装成contig或转录本。然后再使用新的参考转录组进行比对过程,并进行进一步分析。
2.3 新转录本的发现
短读长测序识别新的转录本是 RNA-seq 中最具挑战性的任务之一。目前不少工具如 Cufflinks, iReckon, SLIDE and StringTie 等都可以通过组装手段检测新的转录本,并将他们添加到现有的转录本列表中。由于策略不同,各方法之间通常显示出较大的分歧。
2.4 基因和转录水平的定量
基因和转录本表达水平的量化是RNA-seq 最常见的应用。定量方法主要是基于比对结果,统计比对到每个转录本序列的读数的数量(尽管有一些算法(例如 Sailfish)依赖于读数中的k -mer 计数而不需要经过比对)。 最简单的量化方法是使用 HTSeq-count 或 featureCounts 等程序整合映射read的原始计数。
原始count信息往往需要转化为RPKM(reads per kilobase of exon model per million reads)/ FPKM (fragments per kilobase of exon model per million mapped reads) 以便消除转录本长度和文库大小的影响。此外, TPM (transcripts per million)也被提出,它有效地归一化了分母中转录本组成的差异,而不是简单地除以文库中的read数量,它被认为在不同来源和组成的样本之间更具可比性,但仍可能存在一些偏差。
2.5 差异基因表达分析
差异基因表达分析用于寻找组间显著表达变化的基因,以解释基因表达水平的变化对生物功能的变化。目前差异分析使用最多的统计模型是负二项分布。常用的软件为edgeR,DEseq2。目前,关于差异分析建议在样本足够情况下 RNA-seq 实验至少进行三个生物学重复,以保证结果的重现性和可靠性。
2.6 可变剪接分析
可变剪切在真核生物体内广泛存在,有研究指出,有95%的基因都存在可变剪切现象。可变剪切导致了转录本和蛋白质结构与功能的多态性,是一种重要的转录调控机制。分析可变剪切常用软件包括ASProfile,rMATS,CircSplice,CASH,SGSeq等。 不同软件的可变剪接类型略有不相同,下结果时需要参考软件实际定义。
2.7 基因融合
基因融合是指由于某种机制(如基因组变异)使得两个不同基因的部分序列或全部序列融合到一起,形成了一个新的基因。 基因融合往往是由于染色体易位,中间缺失,染色体倒位等变异形成的。基因融合检测的软件非常多,Nucleic Acids Research杂志也发表过关于这些软件性能评估的论文[4](题为 Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data),感兴趣可以读一读。
参考文献
- 肖慧, 周文浩. 转录组测序在孟德尔遗传病临床诊断中的应用进展[J]. Chinese Journal of Contemporary Pediatrics, 2020, 22(10): 1138.
- Zhang H, He L, Cai L. Transcriptome sequencing: RNA-seq[J]. Computational Systems Biology: Methods and Protocols, 2018: 15-27.
- Conesa, A., Madrigal, P., Tarazona, S. et al. A survey of best practices for RNA-seq data analysis. Genome Biol 17, 13 (2016). https://doi.org/10.1186/s13059-016-0881-8.
- Liu S, Tsai W H, Ding Y, et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data[J]. Nucleic acids research, 2016, 44(5): e47-e47.