R语言多线程加速
通常情况下,R语言只能使用一个线程来进行计算,因此计算的速度及其感人!
最近刚好有一个需求:我有一个参考数据表,里面存放了30万条基因的名称和位置信息,现在我想从里面找到指定的6000个基因的位置信息。
最简单的方法是用两层for循环进行迭代,一分钟写出以下代码:
for (i in 1:nrow(df)){
pos <- 0
for (m in 1:nrow(ref)){
if (ref$geneID[m] == gene$ID[i]){
pos <- ref$pos[m]
}
}
df$pos[i] <- pos
}这段代码是一个双重循环结构,其中外层循环遍历数据框df中的每一行,内层循环遍历参考数据框ref中的每一行。那么也就是说一共要计算290000*6000次。如果是一个线程跑到花儿都谢了。。。。
R语言中如何实现多线程,使计算速度更快?今天分享一个提高效率的神技能。
前言
在R语言中,for循环是一个非常常见的循环结构,它可以用来遍历数据集、进行计算等。
但是,当数据量较大时,使用for循环进行计算会非常耗时,这时我们可以考虑使用多线程来加速计算。
单线程计算
我们首先来看一下单线程的计算速度,下面是一个简单的for循环示例:
# 生成一个长度为100000000的随机向量
x <- rnorm(100000000)
# 计算向量中所有元素的平方
start_time <- Sys.time()
for (i in 1:length(x)) {
x[i] <- x[i] ^ 2
}
end_time <- Sys.time()
# 输出计算时间
cat("单线程计算时间:", end_time - start_time, "\n")运行以上代码,我们可以得到一个长度为100000000的随机向量,并对其中的每个元素进行平方计算。运行结果如下:
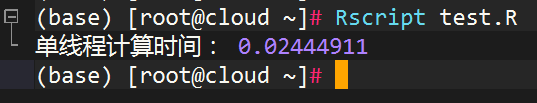
可以看到,单线程计算1000000个元素的平方需要0.024秒左右。
多线程计算
接下来,使用foreach包来实现多线程计算。foreach包是一个非常常用的并行计算包,它可以在多个处理器上并行运行迭代过程。下面是使用foreach包进行多线程计算的示例代码:
# 加载foreach包
library(foreach)
# 生成一个长度为100000的随机向量
x <- rnorm(100000000)
# 使用foreach包进行多线程计算
start_time <- Sys.time()
foreach(i = 1:length(x), .combine = c) %dopar% {
x[i] <- x[i] ^ 2
}
end_time <- Sys.time()
# 输出计算时间
cat("多线程计算时间:", end_time - start_time, "\n")运行以上代码,我们可以得到与单线程相同的结果。但是,由于使用了多线程计算,计算时间会更短。运行结果如下:
多线程计算时间: 0.00099906 可以看到,使用foreach包进行多线程计算100000000个元素的平方只需要0.0009秒左右,比单线程计算快了很多。
多线程计算的原理
在上面的示例中,我们使用了foreach包来实现多线程计算。foreach包的原理是将迭代过程分成多个部分,每个部分在不同的处理器上并行运行。
具体来说,foreach包将迭代过程分成若干个任务,并将这些任务分配给不同的处理器进行计算。每个处理器计算完成后,foreach包会将计算结果合并起来,并返回最终结果。
在上面的示例中,我们使用了%dopar%关键字来表示并行运行。这个关键字告诉foreach包在多个处理器上并行运行迭代过程。另外,我们还使用了.combine = c参数来告诉foreach包如何将计算结果合并起来。这里我们使用了c函数来将计算结果合并成一个向量。
总结
在本文中,介绍了如何在R语言中实现多线程计算,并对比了单线程与多线程的计算速度。可以看到,使用多线程可以大大加快计算速度,特别是在数据量较大时。在实际应用中,我们可以根据自己的需求选择不同的多线程计算包来实现并行计算。
彩蛋
最后,分享本文开始时提出问题的答案,通过以下代码替换原来的双层嵌套for循环,能大幅提高计算速度,从个人使用上来看,原来24小时跑完的任务,现在1小时就能出结果(主要还是CPU核心数限制,不然可能还能再快一点儿)
# 设置并行计算的核心数
num_cores <- detectCores()
cl <- makeCluster(num_cores - 1)
# 注册并行计算集群
registerDoParallel(cl)
# 定义并行计算任务
gene_pos <- foreach(i = 1:nrow(gene), .combine = rbind) %dopar% {
pos <- 0
for (m in 1:nrow(ref)){
if (ref$gene[m] == gene$V1[i]){
pos <- ref$pos[m]
}
}
pos
}
# 结束并行计算
stopCluster(cl)本文由mdnice多平台发布