# WGCNA | 不止一个组的WGCNA怎么分析嘞!?~(四)(共识网络分析-第四步-共识模块与性状相关联)
1写在前面
最近稍微没有那么忙了,好好搞一下公众号吧,好久没怎么认真做了。
有的时候你会发现坏事不一定是坏事,塞翁失马,焉知非福啊!~
"我只担心一件事,我怕我配不上自己所受的苦难。"
慢慢熬吧,毕竟世上无难事,只要肯放弃。
2用到的包
rm(list = ls())
library(tidyverse)
library(WGCNA)
3示例数据
我们这个时候要把前面清洗好,构建好的网络数据拿出来吧。
load("./Consensus-dataInput.RData")
load("./Consensus-NetworkConstruction-auto.RData")
4提取数据集个数
在这里,我们先和之前一样提取一下我们的数据集个数,后面会用到。
exprSize <- checkSets(multiExpr)
nSets <- exprSize$nSets
5计算模块相关性及p值
为了区分共识网络分析结果,我们把变量命名为consMEs、moduleLabels、moduleColors 和 conTree 。
moduleTraitCor = list()
moduleTraitPvalue = list()
for (set in 1:nSets){
moduleTraitCor[[set]] = cor(consMEs[[set]]$data, Traits[[set]]$data, use = "p")
moduleTraitPvalue[[set]] = corPvalueFisher(moduleTraitCor[[set]], exprSize$nSamples[set])
}
6转换label为color
MEColors <- labels2colors(as.numeric(substring(names(consMEs[[1]]$data), 3)))
MEColorNames <- paste("ME", MEColors, sep="")
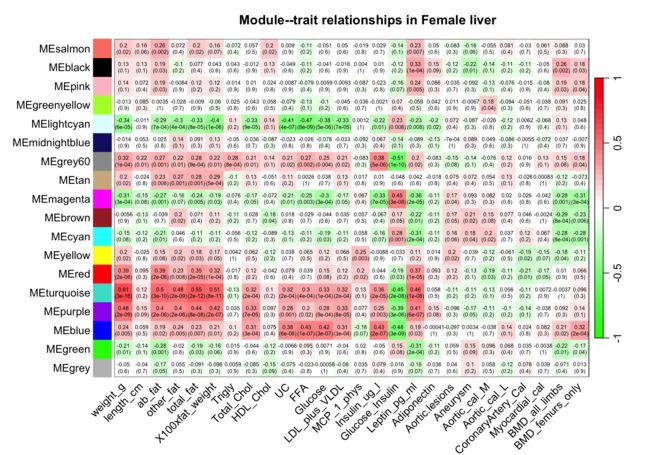
7可视化雌性小鼠module-trait相关性
sizeGrWindow(10,7)
set = 1
textMatrix = paste(signif(moduleTraitCor[[set]], 2), "\n(",
signif(moduleTraitPvalue[[set]], 1), ")", sep = "")
dim(textMatrix) = dim(moduleTraitCor[[set]])
par(mar = c(6, 8.8, 3, 2.2))
labeledHeatmap(Matrix = moduleTraitCor[[set]],
xLabels = names(Traits[[set]]$data),
yLabels = MEColorNames,
ySymbols = MEColorNames,
colorLabels = F,
colors = greenWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = F,
cex.text = 0.5,
zlim = c(-1,1),
main = paste("Module--trait relationships in", setLabels[set]))
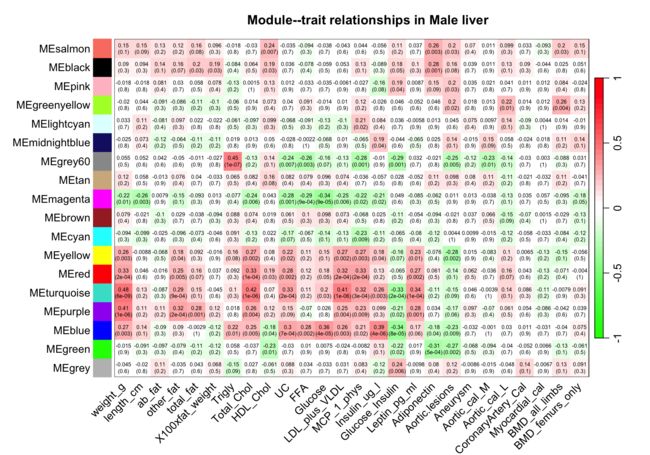
8可视化雄性小鼠module-trait相关性
set = 2
textMatrix = paste(signif(moduleTraitCor[[set]], 2), "\n(",
signif(moduleTraitPvalue[[set]], 1), ")", sep = "")
dim(textMatrix) = dim(moduleTraitCor[[set]])
sizeGrWindow(10,7)
par(mar = c(6, 8.8, 3, 2.2))
labeledHeatmap(Matrix = moduleTraitCor[[set]],
xLabels = names(Traits[[set]]$data),
yLabels = MEColorNames,
ySymbols = MEColorNames,
colorLabels = F,
colors = greenWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = F,
cex.text = 0.5,
zlim = c(-1,1),
main = paste("Module--trait relationships in", setLabels[set]))
9计算共同模块相关性及p值
consensusCor <- matrix(NA, nrow(moduleTraitCor[[1]]), ncol(moduleTraitCor[[1]]))
consensusPvalue <- matrix(NA, nrow(moduleTraitCor[[1]]), ncol(moduleTraitCor[[1]]))
# 找出负相关的
negative <- moduleTraitCor[[1]] < 0 & moduleTraitCor[[2]] < 0
consensusCor[negative] <- pmax(moduleTraitCor[[1]][negative], moduleTraitCor[[2]][negative])
consensusPvalue[negative] <- pmax(moduleTraitPvalue[[1]][negative],
moduleTraitPvalue[[2]][negative])
# 找出正相关的
positive <- moduleTraitCor[[1]] > 0 & moduleTraitCor[[2]] > 0
consensusCor[positive] <- pmin(moduleTraitCor[[1]][positive], moduleTraitCor[[2]][positive])
consensusPvalue[positive] <- pmax(moduleTraitPvalue[[1]][positive],
moduleTraitPvalue[[2]][positive])
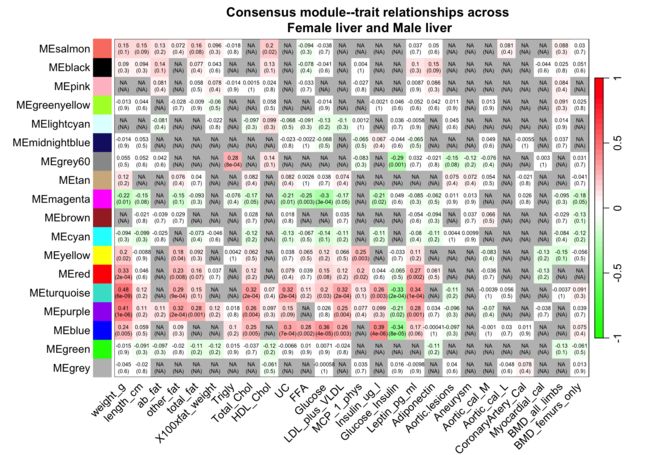
10可视化最终module-trait相关性
textMatrix <- paste(signif(consensusCor, 2), "\n(",
signif(consensusPvalue, 1), ")", sep = "")
dim(textMatrix) <- dim(moduleTraitCor[[set]])
sizeGrWindow(10,7)
par(mar = c(6, 8.8, 3, 2.2))
labeledHeatmap(Matrix = consensusCor,
xLabels = names(Traits[[set]]$data),
yLabels = MEColorNames,
ySymbols = MEColorNames,
colorLabels = F,
colors = greenWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = F,
cex.text = 0.5,
zlim = c(-1,1),
main = paste("Consensus module--trait relationships across\n",
paste(setLabels, collapse = " and ")))
11导出网络文件
11.1 注释探针
file <- gzfile(description = "./FemaleLiver-Data/GeneAnnotation.csv")
annot <- read.csv(file = file)
probes <- names(multiExpr[[1]]$data)
probes2annot <- match(probes, annot$substanceBXH)
11.2 计算gene significances和module memberships
consMEs.unord <- multiSetMEs(multiExpr, universalColors = moduleLabels, excludeGrey = T)
GS = list()
kME = list()
for (set in 1:nSets){
GS[[set]] = corAndPvalue(multiExpr[[set]]$data, Traits[[set]]$data)
kME[[set]] = corAndPvalue(multiExpr[[set]]$data, consMEs.unord[[set]]$data)}

11.3 Z-scores计算相关性
GS.metaZ <- (GS[[1]]$Z + GS[[2]]$Z)/sqrt(2)
kME.metaZ <- (kME[[1]]$Z + kME[[2]]$Z)/sqrt(2)
GS.metaP <- 2*pnorm(abs(GS.metaZ), lower.tail = F)
kME.metaP <- 2*pnorm(abs(kME.metaZ), lower.tail = F)
11.4 整合GS和kME等数据
GSmat <- rbind(GS[[1]]$cor, GS[[2]]$cor, GS[[1]]$p, GS[[2]]$p, GS.metaZ, GS.metaP)
nTraits <- checkSets(Traits)$nGenes
traitNames <- colnames(Traits[[1]]$data)
dim(GSmat) <- c(nGenes, 6*nTraits)
rownames(GSmat) <- probes
colnames(GSmat) <- spaste(c("GS.set1.", "GS.set2.", "p.GS.set1.", "p.GS.set2.", "Z.GS.meta.", "p.GS.meta"), rep(traitNames, rep(6, nTraits)))
kMEmat <- rbind(kME[[1]]$cor, kME[[2]]$cor, kME[[1]]$p, kME[[2]]$p, kME.metaZ, kME.metaP)
MEnames <- colnames(consMEs.unord[[1]]$data)
nMEs <- checkSets(consMEs.unord)$nGenes
dim(kMEmat) <- c(nGenes, 6*nMEs)
rownames(kMEmat) <- probes
colnames(kMEmat) <- spaste(
c("kME.set1.", "kME.set2.", "p.kME.set1.", "p.kME.set2.", "Z.kME.meta.", "p.kME.meta"),
rep(MEnames, rep(6, nMEs)))
11.5 输出文件
info <- data.frame(Probe = probes, GeneSymbol = annot$gene_symbol[probes2annot],
EntrezID = annot$LocusLinkID[probes2annot],
ModuleLabel = moduleLabels,
ModuleColor = labels2colors(moduleLabels),GSmat, kMEmat)
write.csv(info, file = "./consensusAnalysis-CombinedNetworkResults.csv", row.names = F, quote = F)
点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
chatPDF | 别再自己读文献了!让chatGPT来帮你读吧!~
WGCNA | 值得你深入学习的生信分析方法!~
ComplexHeatmap | 颜狗写的高颜值热图代码!
ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
scRNA-seq | 吐血整理的单细胞入门教程
NetworkD3 | 让我们一起画个动态的桑基图吧~
RColorBrewer | 再多的配色也能轻松搞定!~
rms | 批量完成你的线性回归
CMplot | 完美复刻Nature上的曼哈顿图
Network | 高颜值动态网络可视化工具
boxjitter | 完美复刻Nature上的高颜值统计图
linkET | 完美解决ggcor安装失败方案(附教程)
......
本文由 mdnice 多平台发布