从EBI中ENA数据库下载原始数据accession的详细信息以及序列
文章目录
-
-
- 1. ENA 数据库的检索功能
-
-
- 1. 简单搜索
- 2. 复杂搜索
-
- 2. ENA 数据库中的accession的详细信息下载
- 3. ENA 数据库中的accession的序列下载
-
学生物的大概都会用到EBI数据库,而其中的ENA数据库更是包含着丰度的序列信息,那么怎么获取他们呢?
1. ENA 数据库的检索功能
ENA数据库网址:https://www.ebi.ac.uk/ena/browser/home
在下载信息之前首先是检索信息:
1. 简单搜索
如下图,直接搜多sediment的信息,就会有很多相关的序列信息, search 搜索框可以所有蛋白,RNA,DNA,菌株分类名字,accession号等。

2. 复杂搜索
想要你的所有更加精确,则需要使用advanced search。在search一栏的下拉选项中。
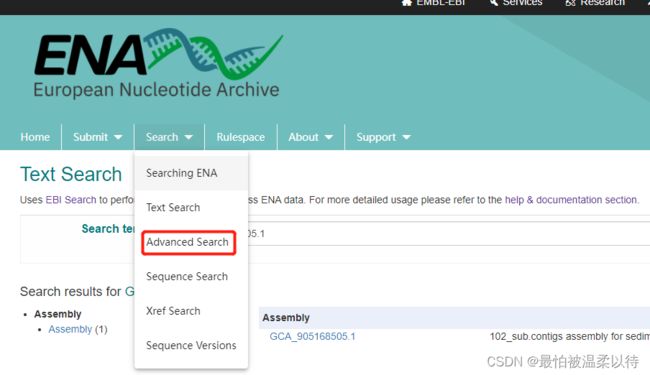
advance search 分为以下这些步骤,其中作为条件筛选的是Data Type 和Query选项。
(Query也可以根据ENA检索的语法规则来写代码检索)
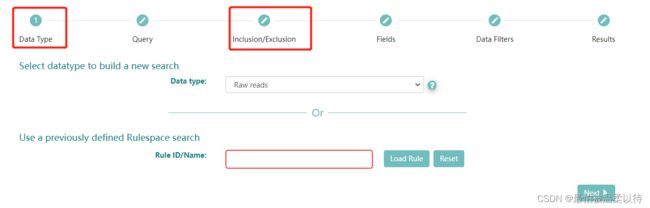
Data Type:是你选择的序列的类型,这里选择Raw reads, 测序的原始数据。Rule ID/Name 不用填。

Query 选项:是你要筛选的序列的信息条件。在左边的对面选项框内选择相应的条件。
例如:我选的是libraray source 和 library strategy 分别是 metagenomic 和 wgs, 也就是说我要筛选所有宏基因组测的全基因组数据。另外在Query框内会生成对应的ENA 数据库的代码。 这里当然也可以在选择其他的条件,最后点击右下方的search 选项就会出现结果了。

所有结果的下载:我后续的处理都是根据下载的txt文件来做的。所以点击TSV下载就可以得到对应的txx文件了。
文件中的结果就是简略的accession号对应的title信息。
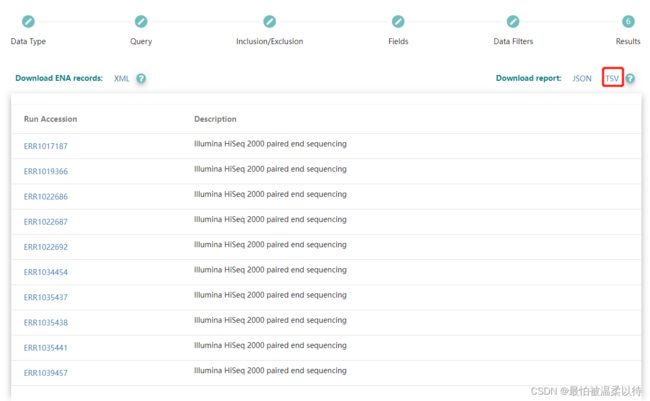
结果文件:

2. ENA 数据库中的accession的详细信息下载
这一部是我自己写的python脚本实现的。直接附上源码。
因为用了递归和异步下载,所以中间在循环时会有网络请求超时等报错信息,不用管他,等程序结束就好了。
程序只需要修改最后最后一步中的文件名就可以用了。
# 需要的包可以自己下载
import aiohttp
import aiofiles
import asyncio
import requests
import pandas as pd
import time
import csv
"""
1. 根据在ENA数据库下载的txt文件,爬取accession的详细信息
2. 生成的结果是多个output文件,然后合并就好了
"""
def read_file(file_name):
with open(file_name, 'r', encoding='utf-8')as f:
f = f.readlines()
accession_list = []
for i in f[1::]:
accession = i.strip().split('\t')[0].strip('"')
accession_list.append(accession)
return accession_list
async def get_biosample_info(accession_number, session, df):
headers = {
# "Referer": f'https://www.ebi.ac.uk/ena/browser/view/{accession_number}',
"User-Agent": "Mozilla/5.0 (Windows NT 10.0; Win64; x64) AppleWebKit/537.36 (KHTML, like Gecko) Chrome/96.0.4664.110 Safari/537.36",
}
params = {
"result": "read_run",
"accession": accession_number,
"offset": 0,
"limit": 1000,
"format": "json",
"fields": "study_accession,secondary_study_accession,sample_accession,secondary_sample_accession,experiment_accession,run_accession,submission_accession,tax_id,scientific_name,instrument_platform,instrument_model,library_name,nominal_length,library_layout,library_strategy,library_source,library_selection,read_count,base_count,center_name,first_public,last_updated,experiment_title,study_title,study_alias,experiment_alias,run_alias,fastq_bytes,fastq_md5,fastq_ftp,fastq_aspera,fastq_galaxy,submitted_bytes,submitted_md5,submitted_ftp,submitted_aspera,submitted_galaxy,submitted_format,sra_bytes,sra_md5,sra_ftp,sra_aspera,sra_galaxy,cram_index_ftp,cram_index_aspera,cram_index_galaxy,sample_alias,broker_name,sample_title,nominal_sdev,first_created"
}
url = r'https://www.ebi.ac.uk/ena/portal/api/filereport'
async with session.get(url, headers=headers, params=params) as resp:
if resp.content:
biosample_info_dic = await resp.json()
biosample_info_dic = biosample_info_dic[0]
print('now dealing with %s'%accession_number)
for k, v in biosample_info_dic.items():
df.loc[accession_number, k] = v
resp.close()
async def main(accession_list, num, input_file):
tasks = []
df = pd.DataFrame()
if accession_list:
# 防止ssl报错
timeout = aiohttp.ClientTimeout(total=600) # 将超时时间设置为600秒
# force_close=True
connector = aiohttp.TCPConnector(limit=80, ssl=False) # 将并发数量降低, 不用ssl验证
async with aiohttp.ClientSession(connector=connector, timeout=timeout) as session:
for accession in accession_list:
task = asyncio.create_task(get_biosample_info(accession, session, df))
tasks.append(task)
await asyncio.wait(tasks)
print(df)
output_file = input_file.split('_')[0]+"_output"+str(num)+".xlsx"
df.to_excel(output_file)
# 找出未下载的accession号
accessions = list(df.index)
not_download_accession = [i for i in accession_list if i not in accessions]
# 递归下载
await main(not_download_accession, int(num)+1, input_file)
if __name__ == '__main__':
try:
print('开始下载')
loop = asyncio.get_event_loop()
# 将下载得到的文件名修改就可以了
loop.run_until_complete(main(read_file('results_read_run_tsv.txt'), 1, 'results_read_run_tsv.txt'))
loop.close()
print("下载结束")
except:
pass
结果就是各个accession对应的详细信息。(需要手动将多个output的结果合并一下就好了,最后懒得写程序合并了)

3. ENA 数据库中的accession的序列下载
在文件中的sra_ftp 和 sra_aspera中会有对应的下载地址,自己把网址整合一下就可以批量下载了。
