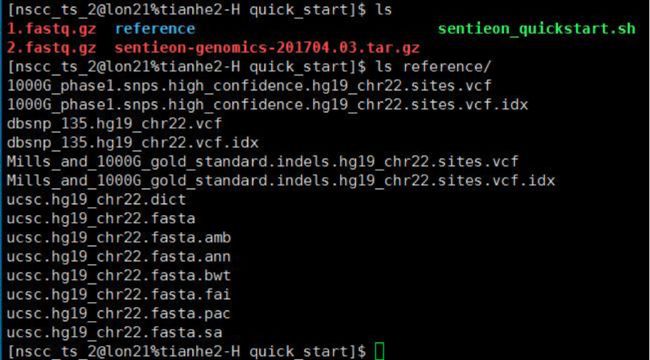
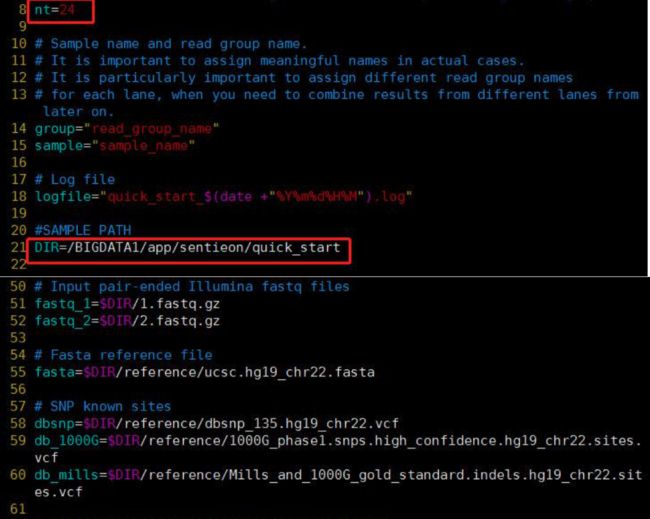
- 机器学习与深度学习间关系与区别
ℒℴѵℯ心·动ꦿ໊ོ꫞
人工智能学习深度学习python
一、机器学习概述定义机器学习(MachineLearning,ML)是一种通过数据驱动的方法,利用统计学和计算算法来训练模型,使计算机能够从数据中学习并自动进行预测或决策。机器学习通过分析大量数据样本,识别其中的模式和规律,从而对新的数据进行判断。其核心在于通过训练过程,让模型不断优化和提升其预测准确性。主要类型1.监督学习(SupervisedLearning)监督学习是指在训练数据集中包含输入
- Goolge earth studio 进阶4——路径修改与平滑
陟彼高冈yu
Googleearthstudio进阶教程旅游
如果我们希望在大约中途时获得更多的城市鸟瞰视角。可以将相机拖动到这里并创建一个新的关键帧。camera_target_clip_7EarthStudio会自动平滑我们的路径,所以当我们通过这个关键帧时,不是一个生硬的角度,而是一个平滑的曲线。camera_target_clip_8路径上有贝塞尔控制手柄,允许我们调整路径的形状。右键单击,我们可以选择“平滑路径”,这是默认的自动平滑算法,或者我们可
- python os.environ_python os.environ 读取和设置环境变量
weixin_39605414
pythonos.environ
>>>importos>>>os.environ.keys()['LC_NUMERIC','GOPATH','GOROOT','GOBIN','LESSOPEN','SSH_CLIENT','LOGNAME','USER','HOME','LC_PAPER','PATH','DISPLAY','LANG','TERM','SHELL','J2REDIR','LC_MONETARY','QT_QPA
- 基于社交网络算法优化的二维最大熵图像分割
智能算法研学社(Jack旭)
智能优化算法应用图像分割算法php开发语言
智能优化算法应用:基于社交网络优化的二维最大熵图像阈值分割-附代码文章目录智能优化算法应用:基于社交网络优化的二维最大熵图像阈值分割-附代码1.前言2.二维最大熵阈值分割原理3.基于社交网络优化的多阈值分割4.算法结果:5.参考文献:6.Matlab代码摘要:本文介绍基于最大熵的图像分割,并且应用社交网络算法进行阈值寻优。1.前言阅读此文章前,请阅读《图像分割:直方图区域划分及信息统计介绍》htt
- 使用 FinalShell 进行远程连接(ssh 远程连接 Linux 服务器)
编程经验分享
开发工具服务器sshlinux
目录前言基本使用教程新建远程连接连接主机自定义命令路由追踪前言后端开发,必然需要和服务器打交道,部署应用,排查问题,查看运行日志等等。一般服务器都是集中部署在机房中,也有一些直接是云服务器,总而言之,程序员不可能直接和服务器直接操作,一般都是通过ssh连接来登录服务器。刚接触远程连接时,使用的是XSHELL来远程连接服务器,连接上就能够操作远程服务器了,但是仅用XSHELL并没有上传下载文件的功能
- 121. 买卖股票的最佳时机
薄荷糖的味道_fb40
给定一个数组,它的第i个元素是一支给定股票第i天的价格。如果你最多只允许完成一笔交易(即买入和卖出一支股票),设计一个算法来计算你所能获取的最大利润。注意你不能在买入股票前卖出股票。示例1:输入:[7,1,5,3,6,4]输出:5解释:在第2天(股票价格=1)的时候买入,在第5天(股票价格=6)的时候卖出,最大利润=6-1=5。注意利润不能是7-1=6,因为卖出价格需要大于买入价格。示例2:输入:
- 每日算法&面试题,大厂特训二十八天——第二十天(树)
肥学
⚡算法题⚡面试题每日精进java算法数据结构
目录标题导读算法特训二十八天面试题点击直接资料领取导读肥友们为了更好的去帮助新同学适应算法和面试题,最近我们开始进行专项突击一步一步来。上一期我们完成了动态规划二十一天现在我们进行下一项对各类算法进行二十八天的一个小总结。还在等什么快来一起肥学进行二十八天挑战吧!!特别介绍小白练手专栏,适合刚入手的新人欢迎订阅编程小白进阶python有趣练手项目里面包括了像《机器人尬聊》《恶搞程序》这样的有趣文章
- 回溯算法-重新安排行程
chirou_
算法数据结构图论c++图搜索
leetcode332.重新安排行程这题我还没自己ac过,只能现在凭着刚学完的热乎劲把我对题解的理解记下来。本题我认为对数据结构的考察比较多,用什么数据结构去存数据,去读取数据,都是很重要的。classSolution{private:unordered_map>targets;boolbacktracking(intticketNum,vector&result){//1.确定参数和返回值//2
- Faiss:高效相似性搜索与聚类的利器
网络·魚
大数据faiss
Faiss是一个针对大规模向量集合的相似性搜索库,由FacebookAIResearch开发。它提供了一系列高效的算法和数据结构,用于加速向量之间的相似性搜索,特别是在大规模数据集上。本文将介绍Faiss的原理、核心功能以及如何在实际项目中使用它。Faiss原理:近似最近邻搜索:Faiss的核心功能之一是近似最近邻搜索,它能够高效地在大规模数据集中找到与给定查询向量最相似的向量。这种搜索是近似的,
- nosql数据库技术与应用知识点
皆过客,揽星河
NoSQLnosql数据库大数据数据分析数据结构非关系型数据库
Nosql知识回顾大数据处理流程数据采集(flume、爬虫、传感器)数据存储(本门课程NoSQL所处的阶段)Hdfs、MongoDB、HBase等数据清洗(入仓)Hive等数据处理、分析(Spark、Flink等)数据可视化数据挖掘、机器学习应用(Python、SparkMLlib等)大数据时代存储的挑战(三高)高并发(同一时间很多人访问)高扩展(要求随时根据需求扩展存储)高效率(要求读写速度快)
- insert into select 主键自增_mybatis拦截器实现主键自动生成
weixin_39521651
insertintoselect主键自增mybatisdelete返回值mybatisinsert返回主键mybatisinsert返回对象mybatisplusinsert返回主键mybatisplus插入生成id
前言前阵子和朋友聊天,他说他们项目有个需求,要实现主键自动生成,不想每次新增的时候,都手动设置主键。于是我就问他,那你们数据库表设置主键自动递增不就得了。他的回答是他们项目目前的id都是采用雪花算法来生成,因此为了项目稳定性,不会切换id的生成方式。朋友问我有没有什么实现思路,他们公司的orm框架是mybatis,我就建议他说,不然让你老大把mybatis切换成mybatis-plus。mybat
- k均值聚类算法考试例题_k均值算法(k均值聚类算法计算题)
寻找你83497
k均值聚类算法考试例题
?算法:第一步:选K个初始聚类中心,z1(1),z2(1),…,zK(1),其中括号内的序号为寻找聚类中心的迭代运算的次序号。聚类中心的向量值可任意设定,例如可选开始的K个.k均值聚类:---------一种硬聚类算法,隶属度只有两个取值0或1,提出的基本根据是“类内误差平方和最小化”准则;模糊的c均值聚类算法:--------一种模糊聚类算法,是.K均值聚类算法是先随机选取K个对象作为初始的聚类
- ES聚合分析原理与代码实例讲解
光剑书架上的书
大厂Offer收割机面试题简历程序员读书硅基计算碳基计算认知计算生物计算深度学习神经网络大数据AIGCAGILLMJavaPython架构设计Agent程序员实现财富自由
ES聚合分析原理与代码实例讲解1.背景介绍1.1问题的由来在大规模数据分析场景中,特别是在使用Elasticsearch(ES)进行数据存储和检索时,聚合分析成为了一个至关重要的功能。聚合分析允许用户对数据集进行细分和分组,以便深入探索数据的结构和模式。这在诸如实时监控、日志分析、业务洞察等领域具有广泛的应用。1.2研究现状目前,ES聚合分析已经成为现代大数据平台的核心组件之一。它支持多种类型的聚
- Python实现简单的机器学习算法
master_chenchengg
pythonpython办公效率python开发IT
Python实现简单的机器学习算法开篇:初探机器学习的奇妙之旅搭建环境:一切从安装开始必备工具箱第一步:安装Anaconda和JupyterNotebook小贴士:如何配置Python环境变量算法初体验:从零开始的Python机器学习线性回归:让数据说话数据准备:从哪里找数据编码实战:Python实现线性回归模型评估:如何判断模型好坏逻辑回归:从分类开始理论入门:什么是逻辑回归代码实现:使用skl
- 推荐算法_隐语义-梯度下降
_feivirus_
算法机器学习和数学推荐算法机器学习隐语义
importnumpyasnp1.模型实现"""inputrate_matrix:M行N列的评分矩阵,值为P*Q.P:初始化用户特征矩阵M*K.Q:初始化物品特征矩阵K*N.latent_feature_cnt:隐特征的向量个数max_iteration:最大迭代次数alpha:步长lamda:正则化系数output分解之后的P和Q"""defLFM_grad_desc(rate_matrix,l
- Linux vi常用命令
fengyehongWorld
Linuxlinux
参考资料viコマンド(vimコマンド)リファレンス目录一.保存系命令二.删除系命令三.移动系命令四.复制粘贴系命令一.保存系命令⏹保存并退出:wq⏹强制保存并退出:wq!⏹退出(文件未编辑):q⏹强制退出(忽略已编辑内容):q!⏹另存为:w新文件名二.删除系命令⏹删除当前行dd⏹清空整个文档gg:移动到文档顶部dG:删除到最后一行ggdG三.移动系命令⏹移动到文档顶部gg⏹移动到文档底部#方式1G
- K近邻算法_分类鸢尾花数据集
_feivirus_
算法机器学习和数学分类机器学习K近邻
importnumpyasnpimportpandasaspdfromsklearn.datasetsimportload_irisfromsklearn.model_selectionimporttrain_test_splitfromsklearn.metricsimportaccuracy_score1.数据预处理iris=load_iris()df=pd.DataFrame(data=ir
- 数据结构 | 栈和队列
TT-Kun
数据结构与算法数据结构栈队列C语言
文章目录栈和队列1.栈:后进先出(LIFO)的数据结构1.1概念与结构1.2栈的实现2.队列:先进先出(FIFO)的数据结构2.1概念与结构2.2队列的实现3.栈和队列算法题3.1有效的括号3.2用队列实现栈3.3用栈实现队列3.4设计循环队列结论栈和队列在计算机科学中,栈和队列是两种基本且重要的数据结构,它们在处理数据存储和访问顺序方面有着独特的规则和应用。本文将详细介绍栈和队列的概念、结构、实
- [Python] 数据结构 详解及代码
AIAdvocate
算法python数据结构链表
今日内容大纲介绍数据结构介绍列表链表1.数据结构和算法简介程序大白话翻译,程序=数据结构+算法数据结构指的是存储,组织数据的方式.算法指的是为了解决实际业务问题而思考思路和方法,就叫:算法.2.算法的5大特性介绍算法具有独立性算法是解决问题的思路和方式,最重要的是思维,而不是语言,其(算法)可以通过多种语言进行演绎.5大特性有输入,需要传入1或者多个参数有输出,需要返回1个或者多个结果有穷性,执行
- Python算法L5:贪心算法
小熊同学哦
Python算法算法python贪心算法
Python贪心算法简介目录Python贪心算法简介贪心算法的基本步骤贪心算法的适用场景经典贪心算法问题1.**零钱兑换问题**2.**区间调度问题**3.**背包问题**贪心算法的优缺点优点:缺点:结语贪心算法(GreedyAlgorithm)是一种在每一步选择中都采取当前最优或最优解的算法。它的核心思想是,在保证每一步局部最优的情况下,希望通过贪心选择达到全局最优解。虽然贪心算法并不总能得到全
- WebMagic:强大的Java爬虫框架解析与实战
Aaron_945
Javajava爬虫开发语言
文章目录引言官网链接WebMagic原理概述基础使用1.添加依赖2.编写PageProcessor高级使用1.自定义Pipeline2.分布式抓取优点结论引言在大数据时代,网络爬虫作为数据收集的重要工具,扮演着不可或缺的角色。Java作为一门广泛使用的编程语言,在爬虫开发领域也有其独特的优势。WebMagic是一个开源的Java爬虫框架,它提供了简单灵活的API,支持多线程、分布式抓取,以及丰富的
- 【RabbitMQ 项目】服务端:数据管理模块之绑定管理
月夜星辉雪
rabbitmq分布式
文章目录一.编写思路二.代码实践一.编写思路定义绑定信息类交换机名称队列名称绑定关键字:交换机的路由交换算法中会用到没有是否持久化的标志,因为绑定是否持久化取决于交换机和队列是否持久化,只有它们都持久化时绑定才需要持久化。绑定就好像一根绳子,两端连接着交换机和队列,当一方不存在,它就没有存在的必要了定义绑定持久化类构造函数:如果数据库文件不存在则创建,打开数据库,创建binding_table插入
- 非对称加密算法原理与应用2——RSA私钥加密文件
私语茶馆
云部署与开发架构及产品灵感记录RSA2048私钥加密
作者:私语茶馆1.相关章节(1)非对称加密算法原理与应用1——秘钥的生成-CSDN博客第一章节讲述的是创建秘钥对,并将公钥和私钥导出为文件格式存储。本章节继续讲如何利用私钥加密内容,包括从密钥库或文件中读取私钥,并用RSA算法加密文件和String。2.私钥加密的概述本文主要基于第一章节的RSA2048bit的非对称加密算法讲述如何利用私钥加密文件。这种加密后的文件,只能由该私钥对应的公钥来解密。
- 粒子群优化 (PSO) 在三维正弦波函数中的应用
subject625Ruben
机器学习人工智能matlab算法
在这篇博客中,我们将展示如何使用粒子群优化(PSO)算法求解三维正弦波函数,并通过增加正弦波扰动,使优化过程更加复杂和有趣。本文将介绍目标函数的定义、PSO参数设置以及算法执行的详细过程,并展示搜索空间中的动态过程和收敛曲线。1.目标函数定义我们使用的目标函数是一个三维正弦波函数,定义如下:objectiveFunc=@(x)sin(sqrt(x(1).^2+x(2).^2))+0.5*sin(5
- 免费的GPT可在线直接使用(一键收藏)
kkai人工智能
gpt
1、LuminAI(https://kk.zlrxjh.top)LuminAI标志着一款融合了星辰大数据模型与文脉深度模型的先进知识增强型语言处理系统,旨在自然语言处理(NLP)的技术开发领域发光发热。此系统展现了卓越的语义把握与内容生成能力,轻松驾驭多样化的自然语言处理任务。VisionAI在NLP界的应用领域广泛,能够胜任从机器翻译、文本概要撰写、情绪分析到问答等众多任务。通过对大量文本数据的
- 非对称加密算法————RSA理论及详情
hu19930613
转自:https://www.kancloud.cn/kancloud/rsa_algorithm/48484一、一点历史1976年以前,所有的加密方法都是同一种模式:(1)甲方选择某一种加密规则,对信息进行加密;(2)乙方使用同一种规则,对信息进行解密。由于加密和解密使用同样规则(简称"密钥"),这被称为"对称加密算法"(Symmetric-keyalgorithm)。这种加密模式有一个最大弱点
- 如何利用大数据与AI技术革新相亲交友体验
h17711347205
回归算法安全系统架构交友小程序
在数字化时代,大数据和人工智能(AI)技术正逐渐革新相亲交友体验,为寻找爱情的过程带来前所未有的变革(编辑h17711347205)。通过精准分析和智能匹配,这些技术能够极大地提高相亲交友系统的效率和用户体验。大数据的力量大数据技术能够收集和分析用户的行为模式、偏好和互动数据,为相亲交友系统提供丰富的信息资源。通过分析用户的搜索历史、浏览记录和点击行为,系统能够深入了解用户的兴趣和需求,从而提供更
- ai绘画工具midjourney怎么下载?附作品管理教程
设计师早上好
Midjourney是一款功能强大的AI绘画工具,它使用机器学习技术和深度神经网络等算法,可以生成各种艺术风格的绘画作品。在创意设计、广告宣传等方面有着广泛的应用前景。那么,ai绘画工具midjourney怎么下载?本文将为您介绍Midjourney的下载以及作品的相关管理。一、Midjourney下载Midjourney的下载非常简单,只需打开Midjourney官网(点击“GetMidjour
- 【加密算法基础——对称加密和非对称加密】
XWWW668899
网络安全服务器笔记
对称加密与非对称加密对称加密和非对称加密是两种基本的加密方法,各自有不同的特点和用途。以下是详细比较:1.对称加密特点密钥:使用相同的密钥进行加密和解密。发送方和接收方必须共享这个密钥。速度:通常速度较快,适合处理大量数据。实现:算法相对简单,计算效率高。常见算法AES(高级加密标准)DES(数据加密标准)3DES(三重数据加密标准)RC4(流密码)应用场景文件加密磁盘加密传输大量数据时的加密2.
- 【算法练习】IDEA集成leetcode插件实现快速刷
2401_84102892
2024年程序员学习算法intellij-idealeetcode
============点击右侧边leetcode->设置->配置地址、用户名、密码、存放目录、文件模板用户名要登录后在账号信息里看模板代码1.codefilename!velocityTool.camelC
- Spring4.1新特性——综述
jinnianshilongnian
spring 4.1
目录
Spring4.1新特性——综述
Spring4.1新特性——Spring核心部分及其他
Spring4.1新特性——Spring缓存框架增强
Spring4.1新特性——异步调用和事件机制的异常处理
Spring4.1新特性——数据库集成测试脚本初始化
Spring4.1新特性——Spring MVC增强
Spring4.1新特性——页面自动化测试框架Spring MVC T
- Schema与数据类型优化
annan211
数据结构mysql
目前商城的数据库设计真是一塌糊涂,表堆叠让人不忍直视,无脑的架构师,说了也不听。
在数据库设计之初,就应该仔细揣摩可能会有哪些查询,有没有更复杂的查询,而不是仅仅突出
很表面的业务需求,这样做会让你的数据库性能成倍提高,当然,丑陋的架构师是不会这样去考虑问题的。
选择优化的数据类型
1 更小的通常更好
更小的数据类型通常更快,因为他们占用更少的磁盘、内存和cpu缓存,
- 第一节 HTML概要学习
chenke
htmlWebcss
第一节 HTML概要学习
1. 什么是HTML
HTML是英文Hyper Text Mark-up Language(超文本标记语言)的缩写,它规定了自己的语法规则,用来表示比“文本”更丰富的意义,比如图片,表格,链接等。浏览器(IE,FireFox等)软件知道HTML语言的语法,可以用来查看HTML文档。目前互联网上的绝大部分网页都是使用HTML编写的。
打开记事本 输入一下内
- MyEclipse里部分习惯的更改
Array_06
eclipse
继续补充中----------------------
1.更改自己合适快捷键windows-->prefences-->java-->editor-->Content Assist-->
Activation triggers for java的右侧“.”就可以改变常用的快捷键
选中 Text
- 近一个月的面试总结
cugfy
面试
本文是在学习中的总结,欢迎转载但请注明出处:http://blog.csdn.net/pistolove/article/details/46753275
前言
打算换个工作,近一个月面试了不少的公司,下面将一些面试经验和思考分享给大家。另外校招也快要开始了,为在校的学生提供一些经验供参考,希望都能找到满意的工作。
- HTML5一个小迷宫游戏
357029540
html5
通过《HTML5游戏开发》摘抄了一个小迷宫游戏,感觉还不错,可以画画,写字,把摘抄的代码放上来分享下,喜欢的同学可以拿来玩玩!
<html>
<head>
<title>创建运行迷宫</title>
<script type="text/javascript"
- 10步教你上传githib数据
张亚雄
git
官方的教学还有其他博客里教的都是给懂的人说得,对已我们这样对我大菜鸟只能这么来锻炼,下面先不玩什么深奥的,先暂时用着10步干净利索。等玩顺溜了再用其他的方法。
操作过程(查看本目录下有哪些文件NO.1)ls
(跳转到子目录NO.2)cd+空格+目录
(继续NO.3)ls
(匹配到子目录NO.4)cd+ 目录首写字母+tab键+(首写字母“直到你所用文件根就不再按TAB键了”)
(查看文件
- MongoDB常用操作命令大全
adminjun
mongodb操作命令
成功启动MongoDB后,再打开一个命令行窗口输入mongo,就可以进行数据库的一些操作。输入help可以看到基本操作命令,只是MongoDB没有创建数据库的命令,但有类似的命令 如:如果你想创建一个“myTest”的数据库,先运行use myTest命令,之后就做一些操作(如:db.createCollection('user')),这样就可以创建一个名叫“myTest”的数据库。
一
- bat调用jar包并传入多个参数
aijuans
下面的主程序是通过eclipse写的:
1.在Main函数接收bat文件传递的参数(String[] args)
如: String ip =args[0]; String user=args[1]; &nbs
- Java中对类的主动引用和被动引用
ayaoxinchao
java主动引用对类的引用被动引用类初始化
在Java代码中,有些类看上去初始化了,但其实没有。例如定义一定长度某一类型的数组,看上去数组中所有的元素已经被初始化,实际上一个都没有。对于类的初始化,虚拟机规范严格规定了只有对该类进行主动引用时,才会触发。而除此之外的所有引用方式称之为对类的被动引用,不会触发类的初始化。虚拟机规范严格地规定了有且仅有四种情况是对类的主动引用,即必须立即对类进行初始化。四种情况如下:1.遇到ne
- 导出数据库 提示 outfile disabled
BigBird2012
mysql
在windows控制台下,登陆mysql,备份数据库:
mysql>mysqldump -u root -p test test > D:\test.sql
使用命令 mysqldump 格式如下: mysqldump -u root -p *** DBNAME > E:\\test.sql。
注意:执行该命令的时候不要进入mysql的控制台再使用,这样会报
- Javascript 中的 && 和 ||
bijian1013
JavaScript&&||
准备两个对象用于下面的讨论
var alice = {
name: "alice",
toString: function () {
return this.name;
}
}
var smith = {
name: "smith",
- [Zookeeper学习笔记之四]Zookeeper Client Library会话重建
bit1129
zookeeper
为了说明问题,先来看个简单的示例代码:
package com.tom.zookeeper.book;
import com.tom.Host;
import org.apache.zookeeper.WatchedEvent;
import org.apache.zookeeper.ZooKeeper;
import org.apache.zookeeper.Wat
- 【Scala十一】Scala核心五:case模式匹配
bit1129
scala
package spark.examples.scala.grammars.caseclasses
object CaseClass_Test00 {
def simpleMatch(arg: Any) = arg match {
case v: Int => "This is an Int"
case v: (Int, String)
- 运维的一些面试题
yuxianhua
linux
1、Linux挂载Winodws共享文件夹
mount -t cifs //1.1.1.254/ok /var/tmp/share/ -o username=administrator,password=yourpass
或
mount -t cifs -o username=xxx,password=xxxx //1.1.1.1/a /win
- Java lang包-Boolean
BrokenDreams
boolean
Boolean类是Java中基本类型boolean的包装类。这个类比较简单,直接看源代码吧。
public final class Boolean implements java.io.Serializable,
- 读《研磨设计模式》-代码笔记-命令模式-Command
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
import java.util.ArrayList;
import java.util.Collection;
import java.util.List;
/**
* GOF 在《设计模式》一书中阐述命令模式的意图:“将一个请求封装
- matlab下GPU编程笔记
cherishLC
matlab
不多说,直接上代码
gpuDevice % 查看系统中的gpu,,其中的DeviceSupported会给出matlab支持的GPU个数。
g=gpuDevice(1); %会清空 GPU 1中的所有数据,,将GPU1 设为当前GPU
reset(g) %也可以清空GPU中数据。
a=1;
a=gpuArray(a); %将a从CPU移到GPU中
onGP
- SVN安装过程
crabdave
SVN
SVN安装过程
subversion-1.6.12
./configure --prefix=/usr/local/subversion --with-apxs=/usr/local/apache2/bin/apxs --with-apr=/usr/local/apr --with-apr-util=/usr/local/apr --with-openssl=/
- sql 行列转换
daizj
sql行列转换行转列列转行
行转列的思想是通过case when 来实现
列转行的思想是通过union all 来实现
下面具体例子:
假设有张学生成绩表(tb)如下:
Name Subject Result
张三 语文 74
张三 数学 83
张三 物理 93
李四 语文 74
李四 数学 84
李四 物理 94
*/
/*
想变成
姓名 &
- MySQL--主从配置
dcj3sjt126com
mysql
linux下的mysql主从配置: 说明:由于MySQL不同版本之间的(二进制日志)binlog格式可能会不一样,因此最好的搭配组合是Master的MySQL版本和Slave的版本相同或者更低, Master的版本肯定不能高于Slave版本。(版本向下兼容)
mysql1 : 192.168.100.1 //master mysq
- 关于yii 数据库添加新字段之后model类的修改
dcj3sjt126com
Model
rules:
array('新字段','safe','on'=>'search')
1、array('新字段', 'safe')//这个如果是要用户输入的话,要加一下,
2、array('新字段', 'numerical'),//如果是数字的话
3、array('新字段', 'length', 'max'=>100),//如果是文本
1、2、3适当的最少要加一条,新字段才会被
- sublime text3 中文乱码解决
dyy_gusi
Sublime Text
sublime text3中文乱码解决
原因:缺少转换为UTF-8的插件
目的:安装ConvertToUTF8插件包
第一步:安装能自动安装插件的插件,百度“Codecs33”,然后按照步骤可以得到以下一段代码:
import urllib.request,os,hashlib; h = 'eb2297e1a458f27d836c04bb0cbaf282' + 'd0e7a30980927
- 概念了解:CGI,FastCGI,PHP-CGI与PHP-FPM
geeksun
PHP
CGI
CGI全称是“公共网关接口”(Common Gateway Interface),HTTP服务器与你的或其它机器上的程序进行“交谈”的一种工具,其程序须运行在网络服务器上。
CGI可以用任何一种语言编写,只要这种语言具有标准输入、输出和环境变量。如php,perl,tcl等。 FastCGI
FastCGI像是一个常驻(long-live)型的CGI,它可以一直执行着,只要激活后,不
- Git push 报错 "error: failed to push some refs to " 解决
hongtoushizi
git
Git push 报错 "error: failed to push some refs to " .
此问题出现的原因是:由于远程仓库中代码版本与本地不一致冲突导致的。
由于我在第一次git pull --rebase 代码后,准备push的时候,有别人往线上又提交了代码。所以出现此问题。
解决方案:
1: git pull
2:
- 第四章 Lua模块开发
jinnianshilongnian
nginxlua
在实际开发中,不可能把所有代码写到一个大而全的lua文件中,需要进行分模块开发;而且模块化是高性能Lua应用的关键。使用require第一次导入模块后,所有Nginx 进程全局共享模块的数据和代码,每个Worker进程需要时会得到此模块的一个副本(Copy-On-Write),即模块可以认为是每Worker进程共享而不是每Nginx Server共享;另外注意之前我们使用init_by_lua中初
- java.lang.reflect.Proxy
liyonghui160com
1.简介
Proxy 提供用于创建动态代理类和实例的静态方法
(1)动态代理类的属性
代理类是公共的、最终的,而不是抽象的
未指定代理类的非限定名称。但是,以字符串 "$Proxy" 开头的类名空间应该为代理类保留
代理类扩展 java.lang.reflect.Proxy
代理类会按同一顺序准确地实现其创建时指定的接口
- Java中getResourceAsStream的用法
pda158
java
1.Java中的getResourceAsStream有以下几种: 1. Class.getResourceAsStream(String path) : path 不以’/'开头时默认是从此类所在的包下取资源,以’/'开头则是从ClassPath根下获取。其只是通过path构造一个绝对路径,最终还是由ClassLoader获取资源。 2. Class.getClassLoader.get
- spring 包官方下载地址(非maven)
sinnk
spring
SPRING官方网站改版后,建议都是通过 Maven和Gradle下载,对不使用Maven和Gradle开发项目的,下载就非常麻烦,下给出Spring Framework jar官方直接下载路径:
http://repo.springsource.org/libs-release-local/org/springframework/spring/
s
- Oracle学习笔记(7) 开发PLSQL子程序和包
vipbooks
oraclesql编程
哈哈,清明节放假回去了一下,真是太好了,回家的感觉真好啊!现在又开始出差之旅了,又好久没有来了,今天继续Oracle的学习!
这是第七章的学习笔记,学习完第六章的动态SQL之后,开始要学习子程序和包的使用了……,希望大家能多给俺一些支持啊!
编程时使用的工具是PLSQL