理论
种群核苷酸多样性,顾名思义指的就是核苷酸多样性,值越大说明核苷酸多样性越高。通常用于衡量群体内的核苷酸多样性,也可以用来推演进化关系。计算公式为:
计算群体的π值,可以理解成先把群体内每个样本两两求解,再求得群体的均值。
计算的软件最常见的是vcftools,也有对应的R包PopGenome。通常是选定某一基因组区域,设定好窗口大小,然后滑动窗口进行计算。
使用VCFTOOLS计算种群核苷酸多样性
VCF文件处理
给VCF文件添加ID
SNP data通常都是以VCF格式文件呈现,老规矩,拿到VCF文件的第一件事情就是添加各个SNP位点的ID。
先看一下最开始生成的VCF文件:
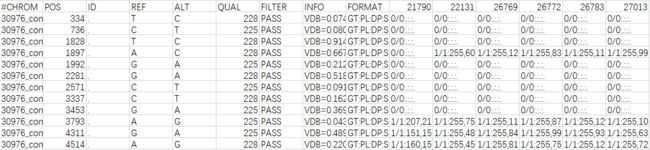
可以看到,ID列都是".",需要我们自己加上去。我用的是某不知名大神写好的perl脚本,可以去我的github上下载(https://github.com/Wanyi-Huang/VCF_add_id),用法:
perl path2file/VCF_add_id.pl YourDataName.vcf YourDataNameWithId.vcf
当然也可以用excel手工添加。添加后的文件如下图所示(格式:CHROMID__POS):
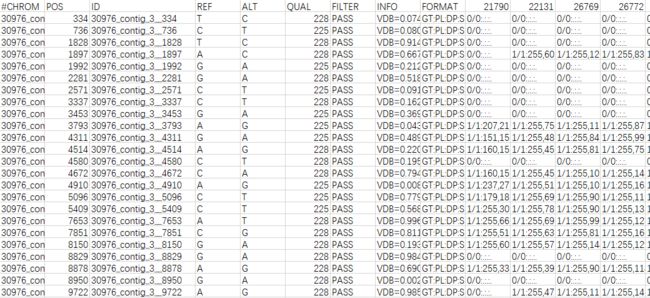
SNP位点过滤
原始Call出来的SNP实在是太多了,而且有一些低频位点会影响后续的分析(软件有时候还会报错),不仅会影响速度,也会影响最后结果的准确性,因此我们去掉他们。此处用到强大的plink软件,用法:
plink --vcf YourDataNameWithId.vcf --maf 0.05 --geno 0.2 --recode vcf-iid -out YourDataNameWithId-maf0.05 --allow-extra-chr
参数解释:--maf 0.05:过滤掉次等位基因频率低于0.05的位点;--geno 0.2:过滤掉有20%的样品缺失的SNP位点;--allow-extra-chr:我的参考数据是Contig级别的,个数比常见分析所用的染色体多太多,所以需要加上此参数。
准备样本ID文件
非常简单,把某群体的全部样本ID放在一个TXT文件里就行,注意要每一行一个ID。
计算核苷酸多样性(π)
vcftools --vcf YourDataNameWithId.vcf --keep YourDataName.txt --window-pi 10000 --out YourDataName_pi
--window-pi 指定窗口的大小,这里我设置了10000,具体大小根据基因组大小选择
--out 指定输出文件的前缀名
数据可视化
数据可视化就是花式展示你的结果。在多样性分析中,π值越大表明群体中该位点的核苷酸多样性越大,反之亦然。那么我们所画的图,应该要展示基因组各个区域π值的大小。因此,我们可以选择散点图or折线图。
画散点图的方法,之前在群体分歧度检验中已经分享过啦,各位移步参考。
那如用R何画折线图嘞?
我的数据集如下图所示:
注:第一列为位置信息;第二列为对应位置的pi值;第三列为需要的颜色;最后一列为染色体位置信息(非必要)。
分享一下我写得一个R流程:(大家需要自己根据自己的数据就行调整,但是万变不离其中,你们可以的!)
#读入数据;
dt1<- read.delim("pi.txt",sep="\t", header = T, check.names = F)
# 加载ggplot2包;
library(ggplot2)
#定义染色体位置。
#我分析的物种有8条染色体,我将所有的染色体都串起来作图,因此需要标出每条染色体的中间位置在哪。
br = c(440000, 1370000, 2410000, 3515000, 4610000,5800000,7095000,8399791)
la = c("1","2","3","4","5","6","7","8")
#自定义图表主题,对图表做精细调整;
mytheme<-theme(panel.grid.major =element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_blank(),
panel.border = element_blank(),
axis.line.y = element_line(color = "black"),
axis.line.x = element_line(color = "black"),
#axis.title.x = element_text(size = rel(1.2)),
axis.title.y = element_text(size = rel(1.2)),
axis.text.y = element_text(size=rel(1.2),color="black"),
#axis.text.x = element_text(size=rel(1.2),color="black"),
plot.margin=unit(x=c(top.mar,right.mar,bottom.mar,left.mar),units="inches"))
#绘制
pa<-ggplot(data=dt1, mapping = aes(x=No,y=pop1))+geom_line(color=dt1$Color1,size=1)
#设置x轴范围,避免点的溢出绘图区;
pa<-pa+scale_x_continuous(limits = c(-1000, 9230000),breaks = br,labels = la)
#设置y轴范围
pa<-pa+scale_y_continuous(limits = c(-0.0005,0.01),breaks = c(0,0.002,0.004,0.006,0.008,0.010),labels = c("0.0","2.0","4.0","6.0","8.0","10.0"))
#设置图例、坐标轴、图表的标题;
pa<-pa+labs(y="Pi (10e-3)",x=NULL)
#自定义图表主题,对图表做精细调整;
pa<-pa+mytheme
#出图
pa
参考:
Vcftools Manual
Comparative genomics revealed adaptive admixture in Cryptosporidium hominis in Africa