- linux服务器上的项目读取本地文件,java访问linux服务器读取文件路径
防晒霜白癜风患者
java访问linux服务器读取文件路径内容精选换一换通过ADC将文件传输到Host。参见准备环境完成环境配置。以运行用户登录安装Toolkit组件的服务器。执行命令,将A.java文件传输到Host的指定路径下。adc--hostxx.xx.xx.xx:22118--sync/tmp/A.java"~/ide_daemon"将xx.xx.xx.xx替换为实际的Host的IP地址。如果Conv2D
- node-imap-sync-client, imap 客户端库, 同步专用
eli960
MAIL前端javascriptnode.js
node-imap-sync-client说明网址:https://gitee.com/linuxmail/node-imap-sync-client同步操作imap客户端,见例子examples本imap客户端,特点:全部命令都是promise风格主要用于和IMAPD服务器同步邮箱数据和邮件数据支持文件夹的创建/删除/移动(改名)支持邮件的复制/移动/删除/标记/上传支持获取文件夹下邮件UID列
- node-ddk, electron 组件,任务栏,托盘,通知
eli960
node-ddkelectronjavascriptnode.js
node-ddk任务栏,托盘,通知https://blog.csdn.net/eli960/article/details/146207062也可以下载demo直接演示http://linuxmail.cn/go#node-ddk在渲染进程(既web端)操作importrenderer,{NODEDDK}from"node-ddk/renderer"letw=renderer.window//让托
- node-ddk,electron 开发组件
eli960
node-ddkelectronjavascript前端node.jsjs
node-ddk-demo说明node-ddk是ELECTRON开发框架,封装常见操作npminode-ddk演示:https://live.csdn.net/v/468440本项目是一个DEMO,项目地址:https://gitee.com/linuxmail/node-ddk-demogitclonehttps://gitee.com/linuxmail/node-ddk-democdnode
- pdm self update 504 gateway timeout
waketzheng
gateway
红军不怕远征难,万里长城今犹在,不见当年秦始皇执行如下命令:pdmselfupdate--verbose时,报了504gatewaytimeout的错误症状:使用的是内网环境的pypimirror,本地Windows有这个问题,服务器Linux系统没有这个问题。经过层层排查,发现是httpx在windows环境读取了注册表里的ProxyServer,但是没有读取ProxyOverride,导致内网
- node-ddk, electron组件, 自定义本地文件协议,打开本地文件
eli960
node-ddkelectronjavascript前端node.js
node-ddk文件协议https://blog.csdn.net/eli960/article/details/146207062也可以下载demo直接演示http://linuxmail.cn/go#node-ddk安全考虑到安全,本系统禁止使用file:///在主窗口,自定义文件协议,可以多个importmain,{NODEDDK}from"node-ddk/main"main.protoc
- conda安装R语言环境并部署至pycharm
楚门留香
r语言开发语言
优先看这个:[win10系统使用Pycharm-professional配置R语言-知乎(zhihu.com)](https://zhuanlan.zhihu.com/p/546788455)要安装R4.0.0的时候看这个:[R语言的安装(详细教程)_r语言安装教程-CSDN博客](https://blog.csdn.net/xhmico/article/details/122443660)r语言
- 区块链环境配置自用
Xmas190
其它区块链
FabricLab1.Fabric环境搭建与基本操作2.Fabric链码基础3.Fabric项目架构Fabric实践一:环境搭建与基本操作一、Fabric环境搭建本文用于指导Fabric在基于Ubuntu的Linux系统中的安装与配置,如有未安装过的同学可以参考本指南自行配置。相关组件版本号:名称版本Ubuntu16.04Fabric1.4Docker20.10.5Docker-compose1.
- RK平台下Buildroot驱动编译环境入门
ItJavawfc
RK系统-驱动驱动学习KernelUbuntuBuildroot
提示:低配置电脑下驱动编译环境搭建,驱动学习环境准备文章目录目的需求环境Ubuntu18Desk桌面开发环境Buildroot编译环境基本要求个人环境VM环境配置+Buildroot编译环境配置Buildroot编译总结目的搭建驱动开发编译环境硬件环境要求不达标如何进行配置规避,使编译环境编译OK为后续自己开发工作中,学习环境做一个简单的指导需求这里我需要搭建的环境是Ubuntu上面用Linux源
- 编译乱序 vs 执行乱序
三境界
操作系统linux驱动开发
背景今天留意了一下linux内核对writel和readl的实现,涉及到了dmb,imb这类屏障指令,过去对这类机制的了解比较模糊,所以查阅了一些资料,做一下记录。#if__LINUX_ARM_ARCH__>=7#defineisb(option)__asm____volatile__("isb"#option:::"memory")#definedsb(option)__asm____volat
- 【R语言2】Introduction to R 基础知识复习小测试 Pop quiz
不二程序猿
r语言开发语言数据挖掘
【R语言】基础知识点Popquiz前言Question1Question2Question3Question4Question5Question6Question7Question8Question9Question10是兄弟就砍一刀!答案前言在这里会有10道题,每一道都是对R语言的基础了解。有单选题和填空题,答案在最下面。填空题可以放到Rstudio里运行得出答案。Question1Whicho
- Qemu&KVM 第一篇 (3)QEMU 架构
weixin_34160277
操作系统
QEMU架构我们首先了解一下QEMU如何实现仿真。本节将介绍QEMU的两种操作模式,以及QEMU动态翻译程序的一些有趣特点。QEMU基本操作QEMU支持两种操作模式:用户模式仿真和系统模式仿真。用户模式仿真允许一个CPU构建的进程在另一个CPU上执行(执行主机CPU指令的动态翻译并相应地转换Linux系统调用)。系统模式仿真允许对整个系统进行仿真,包括处理器和配套的外围设备。在x86主机系统上仿真
- sqlmap笔记
君如尘
网络安全-渗透笔记笔记
1.运行环境sqlmap是用Python编写的,因此首先需要确保你的系统上安装了Python。sqlmap支持Python2.6、2.7和Python3.4及以上版本。2.常用命令通用格式:bythonsqlmap.py-r注入点地址--参数-rpost请求-uget请求--level=测试等级--risk=测试风险-v显示详细信息级别-p针对某个注入点注入-threads更改线程数,加速--ba
- QEMU源码全解析 —— CPU虚拟化(12)
蓝天居士
QEMU/KVMQEMUKVMCPU虚拟化
接前一篇文章:本文内容参考:《趣谈Linux操作系统》——刘超,极客时间《QEMU/KVM》源码解析与应用——李强,机械工业出版社《深度探索Linux系统虚拟化原理与实现》——王柏生谢广军,机械工业出版社特此致谢!三、KVM模块初始化介绍1.KVM简介与源码组织结构KVM全称为Kernel-BasedVirtualMachine,中文译为基于内核的虚拟化技术。KVM是由以色列初创公司Qumrane
- Docker 容器基础技术:namespace
寻雾&启示
docker容器运维
在容器内进程是隔离的,比如容器有自己的网络和文件系统,容器内进程的PID为1,这些都是依赖于Linuxnamespace所提供的隔离机制。本篇我们来了解下Linux有哪些namespace,以及它们是如何实现隔离的。文中案例代码均由ChatGPT生成,在Linux内核5.15.0-124-generic,ubuntu22.04LTS系统上测试通过。namespace类型每个进程都有自己所属的nam
- Linux系统编程:目录操作、文件权限与库管理
网恋东雪莲被骗114514
linux运维服务器
Linux系统编程:目录操作、文件权限与库管理目录的读取在Linux系统编程中,目录操作是常见的任务之一。以下是用于目录操作的核心函数及其用法:1.opendir功能:打开一个目录,返回指向目录流的指针。原型:#includeDIR*opendir(constchar*name);参数:name:目录路径字符串。返回值:成功:返回DIR*指针;失败返回NULL。示例:DIR*dir=opendir
- Python爬虫笔记一(来自MOOC) Requests库入门
小灰不停前进
#Pythonpythonpycharm爬虫
Python爬虫笔记一通用代码框架:importrequestsdefgetHTMLText(url):try:r=requests.get(url,timeput=30)r.raise_for_status()#如果状态不是200,引发HTTPError异常r.encoding=r.apparemt_encodingreturnr.textexcept:return"产生异常"if__name_
- 不神话大模型,不做技术乌托邦,用"传统IT+AI积木"实现企业智能转型
人工智能
一、开篇:AI革命的务实辩证法在技术狂热与落地鸿沟并存的AI时代,灵燕智能体开发平台提出"三轮驱动法则":•不颠覆的智慧:MySQL、知识图谱库、MQ等传统中间件构成数字地基•不空想的创新:大模型仅承担"认知苦力",在人类设计的思考链中定向发力•不取巧的工程:通过D2R映射、低代码工具、元数据治理实现可落地的智能装配二、核心价值:智能开发的工业流水线技术要素原子化拆解将复杂需求分解为可执行的"技术
- Linux脚本实践1
一点多余.
linux运维服务器脚本
前言日常在Liunx中用到多个版本的java修改很麻烦,一个脚本搞定。1.准备两个jdk(如下图所示)2.准备脚本文件viswitch_jdk.sh#!/bin/bash#提示用户输入JDK路径read-p"请输入JDK的绝对路径(例如/usr/local/jdk/jdk-11.0.21):"jdk_path#检查输入的路径是否存在if[!-d"$jdk_path"];thenecho"错误:路径
- Docker之安装与配置
雨五夜
Dockerdocker容器运维
Docker之安装与配置一、Docker环境配置1.基本配置2.镜像加速3.网络配置4.数据持久化5.优化建议6.常见问题与解决方案7.补充工具二、Docker配置本地仓库指南1.拉取Registry镜像2.启动本地仓库3.配置Docker客户端Linux/macOSWindows4.推送镜像到本地仓库标记镜像推送镜像5.推送镜像到本地仓库6.管理本地仓库7.优化与安全性8.常见问题一、Docke
- Linux中的 mutex [二] —— 乐观自旋机制
jianchi88
内核同步Linux稳定性android服务器linuxubuntu
本文基于5.4.86版本内核mutex可视作是spinlock的可睡眠版本,同样是线程无法继续向前执行,但spinlock是"spin",导致该CPU上无法发生线程切换,而mutex是"block"(我们通常翻译成「阻塞」),可以发生线程切换,让所在CPU上的其他线程继续执行。阻塞既可以发生在线程试图获取mutex时,也可以发生在线程持有mutex时。现在的mutex机制,要从这几方面纬度理解:o
- Linux中mutex机制
C嘎嘎嵌入式开发
Linuxlinux运维服务器
在Linux中,mutex是一种用于多线程编程的同步机制,用于保护共享资源,防止多个线程同时访问或修改这些资源,从而避免竞态条件的发生。mutex是“mutualexclusion”的缩写,意为“互斥”。1.Mutex的基本概念互斥锁:mutex是一种锁机制,用于确保在任何时刻只有一个线程可以访问共享资源。当一个线程持有mutex时,其他试图获取该mutex的线程将被阻塞,直到持有mutex的线程
- epoll成员函数介绍
C嘎嘎嵌入式开发
Linux服务器c++开发语言
epoll_create1epoll_create1是Linux系统中用于创建一个新的epoll实例的系统调用。epoll是一种高效的I/O事件通知机制,常用于处理大量的文件描述符(如套接字)。epoll_create1是epoll_create的改进版本,提供了更多的灵活性。函数原型intepoll_create1(intflags);参数说明flags类型:int描述:用于指定创建epoll实
- Linux内核同步机制之(八):mutex
ikt4435
程序员编程Java架构javaspringmysql
一、Mutex锁简介在linux内核中,互斥量(mutex,即mutualexclusion)是一种保证串行化的睡眠锁机制。和spinlock的语义类似,都是允许一个执行线索进入临界区,不同的是当无法获得锁的时候,spinlock原地自旋,而mutex则是选择挂起当前线程,进入阻塞状态。正因为如此,mutex无法在中断上下文使用。和mutex更类似的机制(无法获得锁时都会阻塞)是binarysem
- 嵌入式Linux驱动开发:从基础知识到实践精通
坚持坚持那些年
本文还有配套的精品资源,点击获取简介:嵌入式Linux由于其稳定性、可定制性和丰富资源,在智能设备领域得到广泛应用。掌握嵌入式Linux驱动程序设计对于开发者至关重要。本课程从基础知识点出发,详细介绍了内核接口理解、设备树编程、I/O操作、字符与块设备驱动、网络驱动、电源管理、调试技巧、硬件抽象层、设备模型和模块化编程等关键技能,并通过实际操作实践来强化学习,帮助开发者成长为嵌入式Linux驱动开
- 回归模型评价指标——衡量预测能力
Tang–t
回归数据挖掘人工智能机器学习python
目录一、指标说明1.均方误差(MeanSquaredError,MSE)2.均方根误差(RootMeanSquaredError,RMSE)3.平均绝对误差(MeanAbsoluteError,MAE)4.决定系数(CoefficientofDetermination,R²)5.解释方差(ExplainedVariance,EV)6.最大误差(MaximumError)二、代码一、指标说明回归模型
- pyspark学习rdd处理数据方法——学习记录
亭午
学习
python黑马程序员"""文件,按JSON字符串存储1.城市按销售额排名2.全部城市有哪些商品类别在售卖3.上海市有哪些商品类别在售卖"""frompysparkimportSparkConf,SparkContextimportosimportjsonos.environ['PYSPARK_PYTHON']=r"D:\anaconda\envs\py10\python.exe"#创建Spark
- 回归任务中的评价指标MAE,MSE,RMSE,R-Squared
旺旺棒棒冰
统计学习方法机器学习回归评价指标r2mse
转自博客。仅供自己学习使用,如有侵权,请联系删除分类任务的评价指标有准确率,P值,R值,F1值,而回归任务的评价指标就是MSE,RMSE,MAE、R-SquaredMSE均方误差MSE是真实值与预测值的差值的平方和然后求平均。通过平方的形式便于求导,所以常被用作线性回归的损失函数。MSE=1m∑i=1m(yi−y^i)2MSE=\frac{1}{m}\sum_{i=1}^{m}\left(y_{i
- 国内 npm 镜像源推荐
PyAIGCMaster
我的学习笔记npm前端node.js
国内npm镜像源推荐除了常用的淘宝镜像(https://registry.npmmirror.com),还有以下国内npm镜像源可供选择:1.CNPM(阿里云)地址:https://r.cnpmjs.org/特点:由cnpm提供,支持同步npm官方仓库。提供更快的下载速度和稳定性。使用方法:npmconfigsetregistryhttps://r.cnpmjs.org/2.京东镜像(JFrogA
- linux脚本怎么访问http,如何使用现有的tcp连接从bash脚本访问http服务器?
玲珑阁玉韦
linux脚本怎么访问http
在bashshellscipt中,我使用几个命令行工具(wget,curl,httpie)来测试我的http服务器.当使用例如curl调用GET请求,我看到tcp连接打开到我的服务器并在http通信完成后立即关闭.$curlhttp://10.5.1.1/favicon.ico-o/dev/null为了更好地测试我的服务器的保持活动行为,我想在多个http请求/响应周期中保持tcp连接打开.我可以
- jQuery 跨域访问的三种方式 No 'Access-Control-Allow-Origin' header is present on the reque
qiaolevip
每天进步一点点学习永无止境跨域众观千象
XMLHttpRequest cannot load http://v.xxx.com. No 'Access-Control-Allow-Origin' header is present on the requested resource. Origin 'http://localhost:63342' is therefore not allowed access. test.html:1
- mysql 分区查询优化
annan211
java分区优化mysql
分区查询优化
引入分区可以给查询带来一定的优势,但同时也会引入一些bug.
分区最大的优点就是优化器可以根据分区函数来过滤掉一些分区,通过分区过滤可以让查询扫描更少的数据。
所以,对于访问分区表来说,很重要的一点是要在where 条件中带入分区,让优化器过滤掉无需访问的分区。
可以通过查看explain执行计划,是否携带 partitions
- MYSQL存储过程中使用游标
chicony
Mysql存储过程
DELIMITER $$
DROP PROCEDURE IF EXISTS getUserInfo $$
CREATE PROCEDURE getUserInfo(in date_day datetime)-- -- 实例-- 存储过程名为:getUserInfo-- 参数为:date_day日期格式:2008-03-08-- BEGINdecla
- mysql 和 sqlite 区别
Array_06
sqlite
转载:
http://www.cnblogs.com/ygm900/p/3460663.html
mysql 和 sqlite 区别
SQLITE是单机数据库。功能简约,小型化,追求最大磁盘效率
MYSQL是完善的服务器数据库。功能全面,综合化,追求最大并发效率
MYSQL、Sybase、Oracle等这些都是试用于服务器数据量大功能多需要安装,例如网站访问量比较大的。而sq
- pinyin4j使用
oloz
pinyin4j
首先需要pinyin4j的jar包支持;jar包已上传至附件内
方法一:把汉字转换为拼音;例如:编程转换后则为biancheng
/**
* 将汉字转换为全拼
* @param src 你的需要转换的汉字
* @param isUPPERCASE 是否转换为大写的拼音; true:转换为大写;fal
- 微博发送私信
随意而生
微博
在前面文章中说了如和获取登陆时候所需要的cookie,现在只要拿到最后登陆所需要的cookie,然后抓包分析一下微博私信发送界面
http://weibo.com/message/history?uid=****&name=****
可以发现其发送提交的Post请求和其中的数据,
让后用程序模拟发送POST请求中的数据,带着cookie发送到私信的接入口,就可以实现发私信的功能了。
- jsp
香水浓
jsp
JSP初始化
容器载入JSP文件后,它会在为请求提供任何服务前调用jspInit()方法。如果您需要执行自定义的JSP初始化任务,复写jspInit()方法就行了
JSP执行
这一阶段描述了JSP生命周期中一切与请求相关的交互行为,直到被销毁。
当JSP网页完成初始化后
- 在 Windows 上安装 SVN Subversion 服务端
AdyZhang
SVN
在 Windows 上安装 SVN Subversion 服务端2009-09-16高宏伟哈尔滨市道里区通达街291号
最佳阅读效果请访问原地址:http://blog.donews.com/dukejoe/archive/2009/09/16/1560917.aspx
现在的Subversion已经足够稳定,而且已经进入了它的黄金时段。我们看到大量的项目都在使
- android开发中如何使用 alertDialog从listView中删除数据?
aijuans
android
我现在使用listView展示了很多的配置信息,我现在想在点击其中一条的时候填出 alertDialog,点击确认后就删除该条数据,( ArrayAdapter ,ArrayList,listView 全部删除),我知道在 下面的onItemLongClick 方法中 参数 arg2 是选中的序号,但是我不知道如何继续处理下去 1 2 3
- jdk-6u26-linux-x64.bin 安装
baalwolf
linux
1.上传安装文件(jdk-6u26-linux-x64.bin)
2.修改权限
[root@localhost ~]# ls -l /usr/local/jdk-6u26-linux-x64.bin
3.执行安装文件
[root@localhost ~]# cd /usr/local
[root@localhost local]# ./jdk-6u26-linux-x64.bin&nbs
- MongoDB经典面试题集锦
BigBird2012
mongodb
1.什么是NoSQL数据库?NoSQL和RDBMS有什么区别?在哪些情况下使用和不使用NoSQL数据库?
NoSQL是非关系型数据库,NoSQL = Not Only SQL。
关系型数据库采用的结构化的数据,NoSQL采用的是键值对的方式存储数据。
在处理非结构化/半结构化的大数据时;在水平方向上进行扩展时;随时应对动态增加的数据项时可以优先考虑使用NoSQL数据库。
在考虑数据库的成熟
- JavaScript异步编程Promise模式的6个特性
bijian1013
JavaScriptPromise
Promise是一个非常有价值的构造器,能够帮助你避免使用镶套匿名方法,而使用更具有可读性的方式组装异步代码。这里我们将介绍6个最简单的特性。
在我们开始正式介绍之前,我们想看看Javascript Promise的样子:
var p = new Promise(function(r
- [Zookeeper学习笔记之八]Zookeeper源代码分析之Zookeeper.ZKWatchManager
bit1129
zookeeper
ClientWatchManager接口
//接口的唯一方法materialize用于确定那些Watcher需要被通知
//确定Watcher需要三方面的因素1.事件状态 2.事件类型 3.znode的path
public interface ClientWatchManager {
/**
* Return a set of watchers that should
- 【Scala十五】Scala核心九:隐式转换之二
bit1129
scala
隐式转换存在的必要性,
在Java Swing中,按钮点击事件的处理,转换为Scala的的写法如下:
val button = new JButton
button.addActionListener(
new ActionListener {
def actionPerformed(event: ActionEvent) {
- Android JSON数据的解析与封装小Demo
ronin47
转自:http://www.open-open.com/lib/view/open1420529336406.html
package com.example.jsondemo;
import org.json.JSONArray;
import org.json.JSONException;
import org.json.JSONObject;
impor
- [设计]字体创意设计方法谈
brotherlamp
UIui自学ui视频ui教程ui资料
从古至今,文字在我们的生活中是必不可少的事物,我们不能想象没有文字的世界将会是怎样。在平面设计中,UI设计师在文字上所花的心思和功夫最多,因为文字能直观地表达UI设计师所的意念。在文字上的创造设计,直接反映出平面作品的主题。
如设计一幅戴尔笔记本电脑的广告海报,假设海报上没有出现“戴尔”两个文字,即使放上所有戴尔笔记本电脑的图片都不能让人们得知这些电脑是什么品牌。只要写上“戴尔笔
- 单调队列-用一个长度为k的窗在整数数列上移动,求窗里面所包含的数的最大值
bylijinnan
java算法面试题
import java.util.LinkedList;
/*
单调队列 滑动窗口
单调队列是这样的一个队列:队列里面的元素是有序的,是递增或者递减
题目:给定一个长度为N的整数数列a(i),i=0,1,...,N-1和窗长度k.
要求:f(i) = max{a(i-k+1),a(i-k+2),..., a(i)},i = 0,1,...,N-1
问题的另一种描述就
- struts2处理一个form多个submit
chiangfai
struts2
web应用中,为完成不同工作,一个jsp的form标签可能有多个submit。如下代码:
<s:form action="submit" method="post" namespace="/my">
<s:textfield name="msg" label="叙述:">
- shell查找上个月,陷阱及野路子
chenchao051
shell
date -d "-1 month" +%F
以上这段代码,假如在2012/10/31执行,结果并不会出现你预计的9月份,而是会出现八月份,原因是10月份有31天,9月份30天,所以-1 month在10月份看来要减去31天,所以直接到了8月31日这天,这不靠谱。
野路子解决:假设当天日期大于15号
- mysql导出数据中文乱码问题
daizj
mysql中文乱码导数据
解决mysql导入导出数据乱码问题方法:
1、进入mysql,通过如下命令查看数据库编码方式:
mysql> show variables like 'character_set_%';
+--------------------------+----------------------------------------+
| Variable_name&nbs
- SAE部署Smarty出现:Uncaught exception 'SmartyException' with message 'unable to write
dcj3sjt126com
PHPsmartysae
对于SAE出现的问题:Uncaught exception 'SmartyException' with message 'unable to write file...。
官方给出了详细的FAQ:http://sae.sina.com.cn/?m=faqs&catId=11#show_213
解决方案为:
01
$path
- 《教父》系列台词
dcj3sjt126com
Your love is also your weak point.
你的所爱同时也是你的弱点。
If anything in this life is certain, if history has taught us anything, it is
that you can kill anyone.
不顾家的人永远不可能成为一个真正的男人。 &
- mongodb安装与使用
dyy_gusi
mongo
一.MongoDB安装和启动,widndows和linux基本相同
1.下载数据库,
linux:mongodb-linux-x86_64-ubuntu1404-3.0.3.tgz
2.解压文件,并且放置到合适的位置
tar -vxf mongodb-linux-x86_64-ubun
- Git排除目录
geeksun
git
在Git的版本控制中,可能有些文件是不需要加入控制的,那我们在提交代码时就需要忽略这些文件,下面讲讲应该怎么给Git配置一些忽略规则。
有三种方法可以忽略掉这些文件,这三种方法都能达到目的,只不过适用情景不一样。
1. 针对单一工程排除文件
这种方式会让这个工程的所有修改者在克隆代码的同时,也能克隆到过滤规则,而不用自己再写一份,这就能保证所有修改者应用的都是同一
- Ubuntu 创建开机自启动脚本的方法
hongtoushizi
ubuntu
转载自: http://rongjih.blog.163.com/blog/static/33574461201111504843245/
Ubuntu 创建开机自启动脚本的步骤如下:
1) 将你的启动脚本复制到 /etc/init.d目录下 以下假设你的脚本文件名为 test。
2) 设置脚本文件的权限 $ sudo chmod 755
- 第八章 流量复制/AB测试/协程
jinnianshilongnian
nginxluacoroutine
流量复制
在实际开发中经常涉及到项目的升级,而该升级不能简单的上线就完事了,需要验证该升级是否兼容老的上线,因此可能需要并行运行两个项目一段时间进行数据比对和校验,待没问题后再进行上线。这其实就需要进行流量复制,把流量复制到其他服务器上,一种方式是使用如tcpcopy引流;另外我们还可以使用nginx的HttpLuaModule模块中的ngx.location.capture_multi进行并发
- 电商系统商品表设计
lkl
DROP TABLE IF EXISTS `category`; -- 类目表
/*!40101 SET @saved_cs_client = @@character_set_client */;
/*!40101 SET character_set_client = utf8 */;
CREATE TABLE `category` (
`id` int(11) NOT NUL
- 修改phpMyAdmin导入SQL文件的大小限制
pda158
sqlmysql
用phpMyAdmin导入mysql数据库时,我的10M的
数据库不能导入,提示mysql数据库最大只能导入2M。
phpMyAdmin数据库导入出错: You probably tried to upload too large file. Please refer to documentation for ways to workaround this limit.
- Tomcat性能调优方案
Sobfist
apachejvmtomcat应用服务器
一、操作系统调优
对于操作系统优化来说,是尽可能的增大可使用的内存容量、提高CPU的频率,保证文件系统的读写速率等。经过压力测试验证,在并发连接很多的情况下,CPU的处理能力越强,系统运行速度越快。。
【适用场景】 任何项目。
二、Java虚拟机调优
应该选择SUN的JVM,在满足项目需要的前提下,尽量选用版本较高的JVM,一般来说高版本产品在速度和效率上比低版本会有改进。
J
- SQLServer学习笔记
vipbooks
数据结构xml
1、create database school 创建数据库school
2、drop database school 删除数据库school
3、use school 连接到school数据库,使其成为当前数据库
4、create table class(classID int primary key identity not null)
创建一个名为class的表,其有一
![[GWAS]杂合度质控_第1张图片](http://img.e-com-net.com/image/info8/42924e0e448b446cb93883ccb7492a56.jpg)
![[GWAS]杂合度质控_第2张图片](http://img.e-com-net.com/image/info8/d251755941e84755aff8b9b65f233a98.jpg)
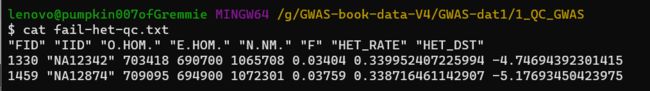
![[GWAS]杂合度质控_第3张图片](http://img.e-com-net.com/image/info8/8495f71fc52e49a59183641bdfe62dc0.jpg)
![[GWAS]杂合度质控_第4张图片](http://img.e-com-net.com/image/info8/7e7869ab90f446f28bd89b51596b28be.jpg)