PDB Database - RCSB PDB 数据集 (2023.8) 的多维度信息统计
欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://spike.blog.csdn.net/article/details/132297736
RCSB PDB 数据集是一个收集了蛋白质的三维结构信息的数据库,是世界蛋白质数据库(wwPDB)的成员之一,也是生物学和医学领域第一个开放访问的数字数据资源库。RCSB PDB 数据集不仅提供了来自蛋白质数据银行(PDB)档案的实验确定的3D结构,还提供了来自 AlphaFold DB 和 ModelArchive 的计算结构模型(CSM)。用户可以利用 RCSB PDB 数据集提供的各种工具和资源,根据序列、结构和功能的注释进行简单和高级搜索,可视化、下载和分析这些分子,并且在外部注释的背景下,探索生物学的结构视角 。
数据维度,数据来源于 RCSB PDB 官网:
pdb_id,即 PDB 的唯一ID,统计 PDB 数量,203657,例如8ETV。chain_id,即从 PDB 中提取的链 ID,例如[A,B,C,D,E,F,I,J],包括蛋白质链与非蛋白质链。file_path,即 PDB 文件路径,pdb8etv.ent.gz,ent表示entry。seq,即序列列表,顺序与chain_id相对应,只包括20个真实氨基酸,不包括扩展氨基酸(protein_letters_3to1_extended)。release_date,发布日期,即1976-05-19~2023-07-26。resolution,即结构分辨率,范围是[0.48,70.0]len,单体或复合物的序列长度,顺序与 chain_id 相对应,例如[75,72,105,95,98,79,110,110],其中,长度统计,len > 20: 201007, len < 20: 2650。max_continuous_missing_res_len,其他指标,来自于 PDB 文件的 head,不太关注。experiment_method,即解析的实验方法,目前已知是13种,例如electron microscopy。chain,真实的链名,来源于 PDB 作者提交,用于匹配之后的数据,与chain_id,可能顺序不同,例如['J', 'I', 'F', 'E', 'D', 'C', 'B', 'A']mol,链的类型,na 表示nucleic acid,即核酸,包括RNA和DNA,protein表示蛋白质,例如['na', 'na', 'protein', 'protein', 'protein', 'protein', 'protein', 'protein'],数量分布{'na': 36136, 'protein': 599619, 'None': 26})。seq_right,来源于 PDB 上传作者提供的序列,与 PDB 结构非对应关系,一般比结构的序列要更长。name,PDB的名称,例如Histone H3.3C,组蛋白。pdb_type,用于表示的蛋白质类型,自定义,例如mul_protein_multimer。num_chain和num_protein_chain,链数与蛋白质的链数,例如8.0、6.0,monomer: 73274, multimer: 115451, sum: 188725。
1. 发布日期 (Release Date)
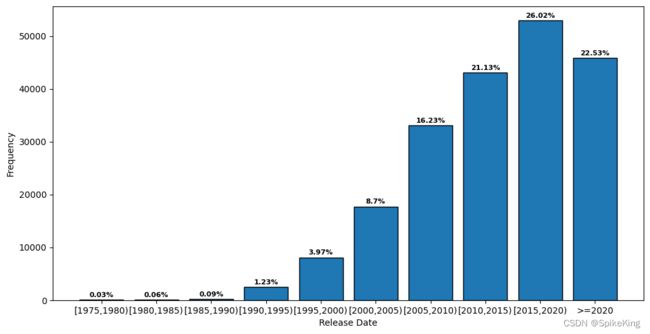
PDB的发布日期(release_date)统计:
- 范围:
1976-05-19~2023-07-26 - 具体间隔 5 年:
1975: 53, 1980: 122, 1985: 190, 1990: 2506, 1995: 8087, 2000: 17725, 2005: 33063, 2010: 43030, 2015: 52988, 2020: 45893, sum: 203657
即:
2. 结构分辨率 (Resolution)
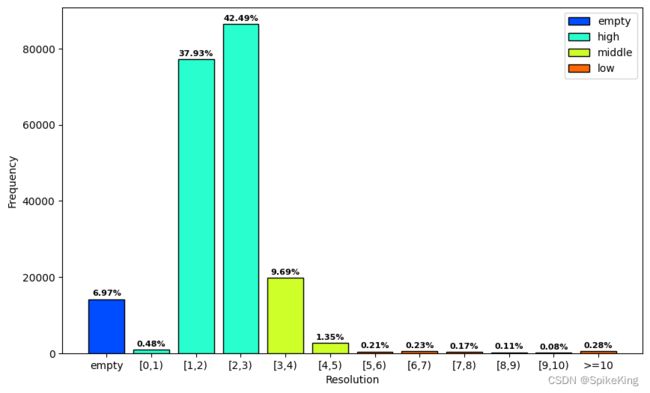
PDB的结构分辨率(resolution)统计:
- 分辨率的范围是
[0.48,70.0] - 具体间隔 1 A:
unknown: 14190, 0: 973, 1: 77252, 2: 86538, 3: 19727, 4: 2758, 5: 437, 6: 463, 7: 355, 8: 232, 9: 165, 10: 567, sum: 203657
即:
3. 蛋白质结构的序列长度 (Seq. Len.)
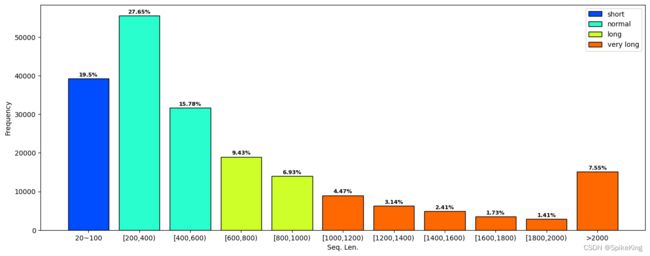
蛋白质的序列长度(Seq. Len.),如果是多聚体 (Multimer),则是多个链的长度之和。
- 序列长度范围:
seq len range: 0 ~ 19350 - 小于20的序列长度:
len > 20: 201007, len < 20: 2650 - 大于20的序列长度:
0: 39201, 200: 55577, 400: 31714, 600: 18964, 800: 13927, 1000: 8989, 1200: 6316, 1400: 4838, 1600: 3468, 1800: 2844, 2000: 15169, sum: 201007
即:
4. 蛋白质结构的解析实验方法 (Experiment Method)
E: 8, EC: 220, EM: 13896, FD: 39, FT: 1, IS: 4, ND: 212, PD: 21, SN: 14117, SS: 78, TM: 7, XD: 175274, sum: 203877,数量203877多于203657,原因是同一个结构,由不同方法解析,SN表示溶液核磁共振与固体核磁共振之和,所以是 12 种方法。- XD (X-ray Diffraction) 是 X射线衍射;SN (Solution/Solid-state NMR) 是核磁共振;EM (Electron Microscopy) 是电镜,也包括冷冻电镜。
- Top3 (XD\ SN \ EM) 占比 99.71%。
即:
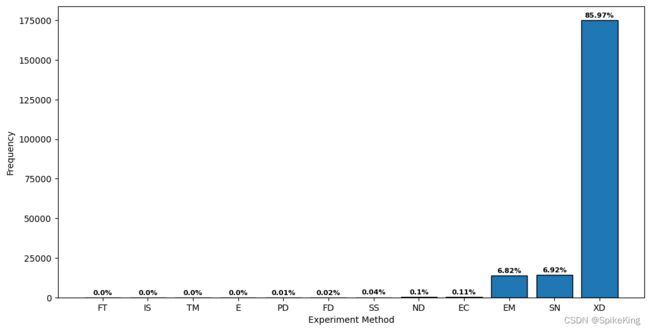
关于实验方法(Experiment Method),一共包括 13 种,同一个蛋白质结构,可以由多种方法共同解析,即:
Electron Crystallography: 电子晶体学
X-ray Diffraction: X射线衍射
Neutron Diffraction: 中子衍射
Solution Scattering: 溶液散射
Solution NMR: 溶液核磁共振
Solid-state NMR: 固体核磁共振
Powder Diffraction: 粉末衍射
Theoretical Model: 理论模型
Fluorescence Transfer: 荧光转移
Infrared Spectroscopy: 红外光谱
Electron Microscopy: 电子显微镜
EPR: 电子顺磁共振
Fiber Diffraction: 纤维衍射
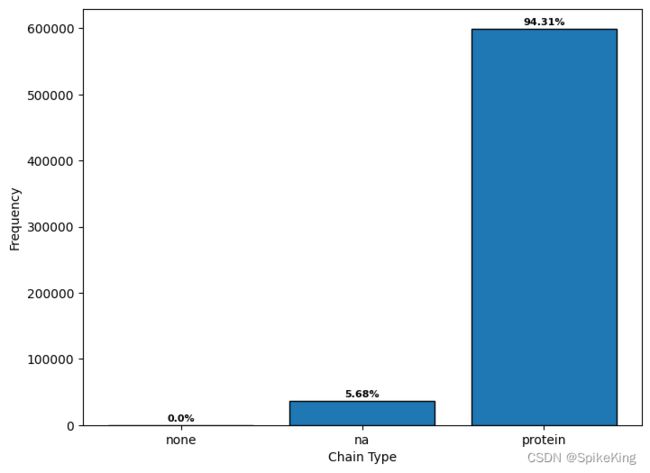
5. 单链类型 (Chain Type)
单链类型主要区分是蛋白质单链,还是核酸 (Nucleic Acid, NA) 单链。
{'na': 36136, 'protein': 599619, 'None': 26}
6. 蛋白质类型 (PDB Name)
提取 PDB Name 中的关键词,去除无意义的标识,例如 数字、subunit、protein、chain、dna、factor、family 等。
- Top 10 的标识:
cytochrome: 2325, dehydrogenase: 3616, glycoprotein: 1978, kinase: 9311, mitochondrial: 1965, polymerase: 3376, protease: 2234, receptor: 6952, reductase: 3746, synthase: 5246
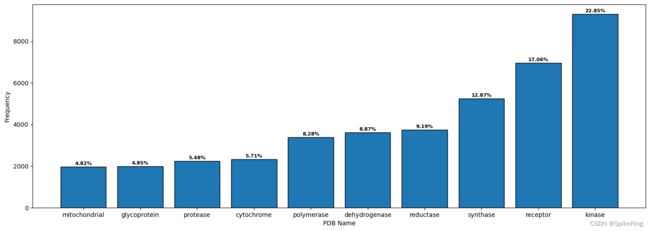
关于 Protein Name 的 Top 10 的标识词,即:
kinase: 激酶
receptor: 受体
synthase: 合酶
reductase: 还原酶
dehydrogenase: 脱氢酶
polymerase: 聚合酶
cytochrome: 细胞色素
protease: 蛋白酶
glycoprotein: 糖蛋白
mitochondrial: 线粒体
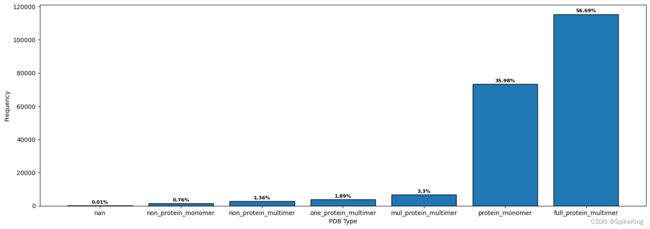
7. 蛋白质链数 (Chain Num)
根据蛋白质链数 (Chain Num),自定义的类型,主要是单体 (Monomer) 和 多聚体 (Multimer),以及根据链的类型,划分成不同类型。
full_protein_multimer: 115451, 全是蛋白质链的 Multimer
protein_monomer: 73274, 蛋白质链的 Monomer
mul_protein_multimer: 6729, 含有 NA 与多个蛋白质链的 Multimer
one_protein_multimer: 3856, 含有 NA 与单个蛋白质链 的 Multimer
non_protein_multimer: 2774, 非蛋白质的 Multimer
non_protein_monomer: 1547, 非蛋白质的 Monomer
nan: 26, 未知
sum: 203657
一致性计算验证,monomer: 73274, multimer: 115451, sum: 188725,数量相同。
8. 压缩数据读取 (ent.gz)
关于 .ent 文件类型的说明:
在 Protein Data Bank 中,包括 .ent 和 .pdb 是两种相同的文件格式,都是用来存储生物大分子的三维结构信息的。ent 是 Protein Data Bank 的原始文件格式,而 pdb 是一种更通用的文件格式,被多种软件识别和处理。ent 和 pdb 的文件内容和结构都是一样的,只是文件扩展名不同而已,可以把 ent 文件的扩展名改为 pdb ,或者用一些转换工具来实现格式转换。ent 表示该文件是一个 entry (条目) 的文件。每个 entry 代表一个生物大分子结构,包含了原子坐标、序列信息、结构注释等数据,每个 entry 都有一个唯一的四位字母或数字代码,称为 PDB ID,例如,1A0A 是人类血红蛋白。
读取 ent.gz 文件,例如 pdb8etv.ent.gz 等。
def read_ent_gz(fpath):
"""
读取 ent gz 文件
"""
with gzip.open(fpath, 'rt', encoding='utf-8') as f:
pdb_str = f.read()
pdb_fh = io.StringIO(pdb_str)
parser = PDBParser(QUIET=True)
structure = parser.get_structure(None, pdb_fh)
header = structure.header
# ...
9. 统计脚本
支持数据统计与图表绘制,调用命令:
python scripts/rcsb_processor.py -i data/pdb_base_info_202308.csv -o mydata/res
即:
#!/usr/bin/env python
# -- coding: utf-8 --
"""
Copyright (c) 2022. All rights reserved.
Created by C. L. Wang on 2023/8/14
"""
import argparse
import ast
import collections
import gzip
import io
import os
import sys
from pathlib import Path
import numpy as np
import pandas as pd
from Bio.PDB import PDBParser
from matplotlib import pyplot as plt
from matplotlib.patches import Rectangle
p = os.path.dirname(os.path.dirname(os.path.abspath(__file__)))
if p not in sys.path:
sys.path.append(p)
from myutils.protein_utils import get_seq_from_pdb
from myutils.project_utils import sort_two_list, sort_dict_by_value, mkdir_if_not_exist
from root_dir import ROOT_DIR
class RcsbProcessor(object):
"""
RCSB数据库统计脚本
"""
def __init__(self):
pass
@staticmethod
def draw_resolution(data_list, save_path=None):
"""
绘制分辨率,分辨率的范围是-1到10,划分11个bin
其中,-1是empty、[1,2,3]是high、其余是low
:param data_list: 数据列表
:param save_path: 存储路径
:return: 绘制图像
"""
labels, counts = np.unique(np.array(data_list), return_counts=True)
labels_str = []
for vl in labels:
if vl == -1:
label = "empty"
else:
label = f"[{vl},{vl+1})"
labels_str.append(label)
labels_str.pop(-1)
labels_str.append(f">={labels[-1]}")
# 颜色设置
cmap = plt.get_cmap('jet')
empty, high, middle, low = cmap(0.2), cmap(0.4), cmap(0.6), cmap(0.8)
color = [empty, high, high, high, middle, middle, low, low, low, low, low, low]
graph = plt.bar(labels_str, counts, align='center', color=color, edgecolor='black')
plt.gca().set_xticks(labels_str)
handles = [Rectangle((0, 0), 1, 1, color=c, ec="k") for c in [empty, high, middle, low]]
color_labels = ["empty", "high", "middle", "low"]
plt.legend(handles, color_labels)
# 绘制百分比
count_sum = sum(counts)
percentage_list = []
for count in counts:
pct = (count / count_sum) * 100
percentage_list.append(round(pct, 2))
i = 0
max_height = max([p.get_height() for p in graph])
for p in graph:
width = p.get_width()
height = p.get_height()
x, y = p.get_xy()
plt.text(x + width / 2,
y + height + max_height*0.01,
str(percentage_list[i]) + '%',
size=8,
ha='center',
weight='bold')
i += 1
# label设置
plt.xlabel("Resolution")
plt.ylabel("Frequency")
# 尺寸以及存储
fig = plt.gcf()
fig.set_size_inches(10, 6)
if save_path:
plt.savefig(save_path, bbox_inches='tight', pad_inches=0.1)
else:
plt.show()
plt.close()
@staticmethod
def draw_release_date(data_list, save_path=None):
"""
绘制发布日期
"""
labels, counts = np.unique(np.array(data_list), return_counts=True)
labels_str = []
for vl in labels:
label = f"[{vl},{vl+5})"
labels_str.append(label)
labels_str.pop(-1)
labels_str.append(f">={labels[-1]}")
# 颜色设置
graph = plt.bar(labels_str, counts, align='center', edgecolor='black')
plt.gca().set_xticks(labels_str)
# 绘制百分比
count_sum = sum(counts)
percentage_list = []
for count in counts:
pct = (count / count_sum) * 100
percentage_list.append(round(pct, 2))
i = 0
max_height = max([p.get_height() for p in graph])
for p in graph:
width = p.get_width()
height = p.get_height()
x, y = p.get_xy()
plt.text(x + width / 2,
y + height + max_height*0.01,
str(percentage_list[i]) + '%',
size=8,
ha='center',
weight='bold')
i += 1
# label设置
plt.xlabel("Release Date")
plt.ylabel("Frequency")
# 尺寸以及存储
fig = plt.gcf()
fig.set_size_inches(12, 6)
if save_path:
plt.savefig(save_path, bbox_inches='tight', pad_inches=0.1)
else:
plt.show()
plt.close()
@staticmethod
def draw_seq_len(data_list, save_path=None):
"""
绘制序列长度
:param data_list: 序列数据集
:param save_path: 图像存储
:return: None
"""
labels, counts = np.unique(np.array(data_list), return_counts=True)
labels_str = []
for vl in labels:
if vl == -1:
label = "empty"
else:
label = f"[{vl},{vl+200})"
labels_str.append(label)
labels_str[-1] = f">{labels[-1]}"
labels_str[0] = f"20~100"
counts = list(counts)
# label设置
plt.xlabel("Seq. Len.")
plt.ylabel("Frequency")
# 颜色设置
cmap = plt.get_cmap('jet')
short, normal, long, v_long = cmap(0.2), cmap(0.4), cmap(0.6), cmap(0.8)
color = [short, normal, normal, long, long, v_long, v_long, v_long, v_long, v_long, v_long]
graph = plt.bar(labels_str, counts, align='center', color=color, edgecolor='black')
plt.gca().set_xticks(labels_str)
handles = [Rectangle((0, 0), 1, 1, color=c, ec="k") for c in [short, normal, long, v_long]]
color_labels = ["short", "normal", "long", "very long"]
plt.legend(handles, color_labels)
# 绘制百分比
count_sum = sum(counts)
percentage_list = []
for count in counts:
pct = (count / count_sum) * 100
percentage_list.append(round(pct, 2))
i = 0
max_height = max([p.get_height() for p in graph])
for p in graph:
width = p.get_width()
height = p.get_height()
x, y = p.get_xy()
plt.text(x + width / 2,
y + height + max_height*0.01,
str(percentage_list[i]) + '%',
size=8,
ha='center',
weight='bold')
i += 1
# 尺寸以及存储
fig = plt.gcf()
fig.set_size_inches(16, 6)
if save_path:
plt.savefig(save_path, bbox_inches='tight', pad_inches=0.1)
else:
plt.show()
plt.close()
@staticmethod
def draw_norm_bars(data_list, x_label, figure_size, save_path=None):
"""
绘制通用的柱状图
"""
labels, counts = np.unique(np.array(data_list), return_counts=True)
counts, labels = sort_two_list(counts, labels)
# 颜色设置
graph = plt.bar(labels, counts, align='center', edgecolor='black')
plt.gca().set_xticks(labels)
# 绘制百分比
count_sum = sum(counts)
percentage_list = []
for count in counts:
pct = (count / count_sum) * 100
percentage_list.append(round(pct, 2))
i = 0
max_height = max([p.get_height() for p in graph])
for p in graph:
width = p.get_width()
height = p.get_height()
x, y = p.get_xy()
plt.text(x + width / 2,
y + height + max_height*0.01,
str(percentage_list[i]) + '%',
size=8,
ha='center',
weight='bold')
i += 1
# label设置
plt.xlabel(x_label)
plt.ylabel("Frequency")
# 尺寸以及存储
fig = plt.gcf()
fig.set_size_inches(*figure_size)
if save_path:
plt.savefig(save_path, bbox_inches='tight', pad_inches=0.1)
else:
plt.show()
plt.close()
@staticmethod
def show_value_counts(data_list):
"""
显示数据统计量
"""
labels, counts = np.unique(np.array(data_list), return_counts=True)
label_res_str = ""
for label, count in zip(labels, counts):
label_res_str += f"{label}: {count}, "
label_res_str = label_res_str[:-2]
print(f"[Info] value_counts: {label_res_str}, sum: {sum(counts)}")
def process_resolution(self, df, output_dir):
"""
统计分辨率
"""
mkdir_if_not_exist(output_dir)
df_resolution_unique = df["resolution"].unique()
df_resolution_unique = sorted(df_resolution_unique)
print(f"[Info] resolution range: [{df_resolution_unique[0]},{df_resolution_unique[-1]}]")
df_resolution = df["resolution"].fillna(-1).astype(int)
df_resolution[df_resolution >= 10] = 10
self.show_value_counts(df_resolution)
self.draw_resolution(df_resolution, os.path.join(output_dir, "resolution.png"))
def process_release_date(self, df, output_dir):
"""
统计发布日期
"""
mkdir_if_not_exist(output_dir)
df_release_date = df["release_date"].unique()
df_release_date = sorted(df_release_date)
print(f"[Info] release_date {df_release_date[0]} - {df_release_date[-1]}")
df["release_date_year"] = df["release_date"].apply(lambda x: int(str(x).split("-")[0]) // 5 * 5)
df_release_date_year = df["release_date_year"]
self.show_value_counts(df_release_date_year)
self.draw_release_date(df_release_date_year, os.path.join(output_dir, "release_date.png"))
def process_seq_len(self, df, output_dir):
"""
统计序列长度
"""
def func(x):
if not isinstance(x, str):
return 0
return sum([int(i) for i in str(x).split(",")])
df["len"] = df["len"].apply(lambda x: func(x))
df_len_unique = df["len"].unique()
df_len_unique = sorted(df_len_unique)
print(f"[Info] seq len range: {df_len_unique[0]} ~ {df_len_unique[-1]}")
df_len_all = df.loc[df['len'] >= 20]
print(f"[Info] len > 20: {len(df_len_all)}, len < 20: {len(df.loc[df['len'] < 20])}")
df_len = df_len_all["len"].astype(int)
df_len[df_len >= 2000] = 2000
df_len = (df_len / 200).astype(int)
df_len = (df_len * 200).astype(int)
self.show_value_counts(df_len)
self.draw_seq_len(df_len, os.path.join(output_dir, "seq_len.png"))
def process_experiment_method(self, df, output_dir):
"""
统计实验方法
"""
experiment_method = df["experiment_method"]
data_list = []
for ex_item in experiment_method:
items = ex_item.split(";")
for item in items:
method = item.strip()
sub_names = method.split(" ")
sub_m = "".join([i[0].upper() for i in sub_names])
data_list.append(sub_m)
self.show_value_counts(data_list)
self.draw_norm_bars(
data_list=data_list, x_label="Experiment Method", figure_size=(12, 6),
save_path=os.path.join(output_dir, "experiment_method.png"))
def process_chain_type(self, df, output_dir):
"""
统计链的类型
"""
df_chain_type = df["mol"]
data_list = []
chain_type_dict = collections.defaultdict(int)
for ct_item in df_chain_type:
if not isinstance(ct_item, str):
data_list.append("none")
chain_type_dict["none"] += 1
continue
ct_item = ast.literal_eval(ct_item)
for item in ct_item:
c_type = item.strip()
chain_type_dict[c_type] += 1
data_list.append(c_type)
print(f"[Info] chain_type: {chain_type_dict}")
self.draw_norm_bars(
data_list=data_list, x_label="Chain Type", figure_size=(8, 6),
save_path=os.path.join(output_dir, "chain_type.png"))
def process_pdb_name(self, df, output_dir):
"""
统计 PDB 名称
"""
df_name = df["name"]
name_dict = collections.defaultdict(int)
for item_str in df_name:
if not isinstance(item_str, str):
name_dict["none"] += 1
continue
items = item_str.split(" ")
for item in items:
name = item.strip().lower()
if not name:
continue
if name in ["subunit", "protein", "chain", "dna", "factor",
"alpha", "family", "light", "heavy", "class",
"putative", "uncharacterized", "domain", "domain-containing", "member",
"hypothetical", "channel"]:
continue
if len(name) <= 4:
continue
name_dict[name] += 1
name_dict_data = sort_dict_by_value(name_dict)
data_list = []
for item in name_dict_data[:10]:
for i in range(item[1]):
data_list.append(item[0])
self.show_value_counts(data_list)
self.draw_norm_bars(
data_list=data_list, x_label="PDB Name", figure_size=(18, 6),
save_path=os.path.join(output_dir, "pdb_name.png"))
def process_pdb_type(self, df, output_dir):
"""
统计 PDB 的类型,主要是 Monomer 与 Multimer,自定义
"""
data_list = list(df["pdb_type"])
self.show_value_counts(data_list)
self.draw_norm_bars(
data_list=data_list, x_label="PDB Type", figure_size=(18, 6),
save_path=os.path.join(output_dir, "pdb_type.png"))
def process_chain_num(self, df, output_dir):
"""
统计链数
"""
num_chain_list = list(df["num_chain"])
num_protein_chain_list = list(df["num_protein_chain"])
data_list = []
for num1, num2 in zip(num_chain_list, num_protein_chain_list):
if num1 != num2:
continue
if num2 > 1:
data_list.append("multimer")
elif num2 == 1:
data_list.append("monomer")
self.show_value_counts(data_list)
self.draw_norm_bars(
data_list=data_list, x_label="Chain Num", figure_size=(6, 6),
save_path=os.path.join(output_dir, "chain_num.png"))
@staticmethod
def read_ent_gz(fpath):
"""
读取 ent gz 文件
"""
with gzip.open(fpath, 'rt', encoding='utf-8') as f:
pdb_str = f.read()
pdb_fh = io.StringIO(pdb_str)
parser = PDBParser(QUIET=True)
structure = parser.get_structure(None, pdb_fh)
model = list(structure.get_models())[0]
# header = structure.header
chain_ids = []
for chain in model:
chain_ids.append(chain.id)
print(f"[Info] chain_id: {chain_ids}")
# ...
def process_profiling(self, csv_path, output_dir):
"""
处理数据库文件
"""
assert os.path.isfile(csv_path)
np.random.seed(42)
print(f"[Info] csv文件: {csv_path}")
df = pd.read_csv(csv_path)
print(df.info())
df_pdb = df["pdb_id"].unique()
rand_idx = np.random.randint(0, len(df_pdb))
print(f"[Info] pdb num: {len(df_pdb)}, {df_pdb[rand_idx]}")
print(f"[Info] chain id: {df['chain_id'][rand_idx]}")
print(f"[Info] file path: {df['filepath'][rand_idx]}")
# --------------- 测试 PDB 文件 --------------- #
pdb_path = os.path.join(ROOT_DIR, "data", "pdb8etv.ent.gz")
self.read_ent_gz(pdb_path)
pdb_path = os.path.join(ROOT_DIR, "data", "pdb8etv.pdb")
seq_str, n_chains, chain_dict = get_seq_from_pdb(pdb_path)
print(f"[Info] protein chain ids: {chain_dict.keys()}")
# --------------- 测试 PDB 文件 --------------- #
seq = df["seq"][rand_idx]
print(f"[Info] seq: {seq}") # 序列
seq_list = seq.split(",")
true_seq_list = list(chain_dict.values())
n_chain = len(true_seq_list)
assert seq_list[0] == true_seq_list[0] and seq_list[n_chain - 1] == true_seq_list[n_chain - 1]
seq_len = df["len"][rand_idx]
print(f"[Info] len: {seq_len}") # 序列长度
max_continuous_missing_res_len = df["max_continuous_missing_res_len"][rand_idx]
print(f"[Info] max_continuous_missing_res_len: {max_continuous_missing_res_len}")
experiment_method = df["experiment_method"][rand_idx]
print(f"[Info] experiment_method: {experiment_method}") # 序列长度
chain_names = df["chain"][rand_idx]
print(f"[Info] chain: {chain_names}")
mol = df["mol"][rand_idx]
print(f"[Info] mol: {mol}")
seq_right = df["seq_right"][rand_idx]
print(f"[Info] seq_right: {seq_right}") # 序列
name = df["name"][rand_idx]
print(f"[Info] name: {name}")
pdb_type = df["pdb_type"][rand_idx]
print(f"[Info] pdb_type: {pdb_type}")
num_chain = df["num_chain"][rand_idx]
num_protein_chain = df["num_protein_chain"][rand_idx]
print(f"[Info] num_chain: {num_chain}, num_protein_chain: {num_protein_chain}")
self.process_release_date(df, output_dir) # 处理发布日期
self.process_resolution(df, output_dir) # 处理分辨率
self.process_seq_len(df, output_dir) # 处理序列长度
self.process_experiment_method(df, output_dir) # 实验方法
self.process_chain_type(df, output_dir) # 实验方法
self.process_pdb_name(df, output_dir) # PDB Name
self.process_pdb_type(df, output_dir) # PDB Type
self.process_chain_num(df, output_dir) # 判断 Monomer 或者 Multimer
def main():
parser = argparse.ArgumentParser()
parser.add_argument(
"-i",
"--input-file",
help="the input file of pdb database profile.",
type=Path,
required=True,
)
parser.add_argument(
"-o",
"--output-dir",
help="the output dir of charts.",
type=Path,
required=True
)
# rcsb_csv_path = os.path.join(ROOT_DIR, "data", "pdb_base_info_202308.csv")
# output_dir = os.path.join(DATA_DIR, "res")
args = parser.parse_args()
input_file = str(args.input_file)
output_dir = str(args.output_dir)
mkdir_if_not_exist(output_dir)
assert os.path.isfile(input_file) and os.path.isdir(output_dir)
rp = RcsbProcessor()
rp.process_profiling(input_file, output_dir)
print("[Info] 全部处理完成! ")
if __name__ == '__main__':
main()
输出日志:
[Info] csv文件: data/pdb_base_info_202308.csv
<class 'pandas.core.frame.DataFrame'>
RangeIndex: 203657 entries, 0 to 203656
Data columns (total 17 columns):
# Column Non-Null Count Dtype
--- ------ -------------- -----
0 Unnamed: 0 203657 non-null int64
1 pdb_id 203657 non-null object
2 chain_id 203574 non-null object
3 resolution 189467 non-null float64
4 release_date 203657 non-null object
5 seq 203574 non-null object
6 len 203574 non-null object
7 max_continuous_missing_res_len 203574 non-null object
8 experiment_method 203657 non-null object
9 chain 203631 non-null object
10 mol 203631 non-null object
11 seq_right 203631 non-null object
12 name 203631 non-null object
13 pdb_type 203631 non-null object
14 num_chain 203631 non-null float64
15 num_protein_chain 203631 non-null float64
16 filepath 203657 non-null object
dtypes: float64(3), int64(1), object(13)
memory usage: 26.4+ MB
[Info] pdb num: 203657, 8etv
[Info] chain id: A,B,C,D,E,F,I,J
[Info] file path: /nfs_beijing_ai/pdb_origin_data/v2023.08/structures/et/pdb8etv.ent.gz
[Info] chain_id: ['A', 'B', 'C', 'D', 'E', 'F', 'I', 'J']
[Info] protein chain ids: dict_keys(['A', 'B', 'C', 'D', 'E', 'F'])
[Info] seq: LLIRKLPFQRLVREIAQDFKTDLRFQSSAVMALQEASEAYLVALFEDTNLAAIHAKRVTIMPKDIQLARRIRGER,RDNIQGITKPAIRRLARRGGVKRISGLIYEETRGVLKVFLENVIRDAVTYTEHAKRKTVTAMDVVYALKRQG,KTRSSRAGLQFPVGRVHRLLRKGNYAERVGAGAPVYLAAVLEYLTAEILELAGNAARDNKKTRIIPRHLQLAVRNDEELNKLLGRVTIAQGGVLPNIQSVLLPKK,KTRKESYAIYVYKVLKQVHPDTGISSKAMSIMNSFVNDVFERIAGEASRLAHYNKRSTITSREIQTAVRLLLPGELAKHAVSEGTKAVTKYTSAK,PHRYRPGTVALREIRRYQKSTELLIRKLPFQRLVREIAQDFKTDLRFQSSAVMALQEASEAYLVALFEDTNLAAIHAKRVTIMPKDIQLARRIRGERA,DNIQGITKPAIRRLARRGGVKRISGLIYEETRGVLKVFLENVIRDAVTYTEHAKRKTVTAMDVVYALKRQGRTLYGFGG,XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX,XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
[Info] len: 75,72,105,95,98,79,110,110
[Info] max_continuous_missing_res_len: 0,0,0,0,0,0,0,0
[Info] experiment_method: electron microscopy
[Info] chain: ['J', 'I', 'F', 'E', 'D', 'C', 'B', 'A']
[Info] mol: ['na', 'na', 'protein', 'protein', 'protein', 'protein', 'protein', 'protein']
[Info] seq_right: ['GGGAGTAATCCCCTTGGCGGTTAAAACGCGGGGGACAGCGCGTACGTGCGTTTAAGCGGTGCTAGAGCTGTCTACGACCAATTGAGCGGCCTCGGCACCGGGATTCTCCA', 'TGGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTACTCCC', 'DNIQGITKPAIRRLARRGGVKRISGLIYEETRGVLKVFLENVIRDAVTYTEHAKRKTVTAMDVVYALKRQGRTLYGFGG', 'PHRYRPGTVALREIRRYQKSTELLIRKLPFQRLVREIAQDFKTDLRFQSSAVMALQEASEAYLVALFEDTNLAAIHAKRVTIMPKDIQLARRIRGERA', 'KTRKESYAIYVYKVLKQVHPDTGISSKAMSIMNSFVNDVFERIAGEASRLAHYNKRSTITSREIQTAVRLLLPGELAKHAVSEGTKAVTKYTSAK', 'KTRSSRAGLQFPVGRVHRLLRKGNYAERVGAGAPVYLAAVLEYLTAEILELAGNAARDNKKTRIIPRHLQLAVRNDEELNKLLGRVTIAQGGVLPNIQSVLLPKK', 'RDNIQGITKPAIRRLARRGGVKRISGLIYEETRGVLKVFLENVIRDAVTYTEHAKRKTVTAMDVVYALKRQG', 'LLIRKLPFQRLVREIAQDFKTDLRFQSSAVMALQEASEAYLVALFEDTNLAAIHAKRVTIMPKDIQLARRIRGER']
[Info] name: Histone H3.3C
[Info] pdb_type: mul_protein_multimer
[Info] num_chain: 8.0, num_protein_chain: 6.0
[Info] release_date 1976-05-19 - 2023-07-26
[Info] value_counts: 1975: 53, 1980: 122, 1985: 190, 1990: 2506, 1995: 8087, 2000: 17725, 2005: 33063, 2010: 43030, 2015: 52988, 2020: 45893, sum: 203657
[Info] resolution range: [0.48,70.0]
[Info] value_counts: -1: 14190, 0: 973, 1: 77252, 2: 86538, 3: 19727, 4: 2758, 5: 437, 6: 463, 7: 355, 8: 232, 9: 165, 10: 567, sum: 203657
[Info] seq len range: 0 ~ 19350
[Info] len > 20: 201007, len < 20: 2650
[Info] value_counts: 0: 39201, 200: 55577, 400: 31714, 600: 18964, 800: 13927, 1000: 8989, 1200: 6316, 1400: 4838, 1600: 3468, 1800: 2844, 2000: 15169, sum: 201007
[Info] value_counts: E: 8, EC: 220, EM: 13896, FD: 39, FT: 1, IS: 4, ND: 212, PD: 21, SN: 14117, SS: 78, TM: 7, XD: 175274, sum: 203877
[Info] chain_type: defaultdict(<class 'int'>, {'na': 36136, 'protein': 599619, 'none': 26})
[Info] value_counts: cytochrome: 2325, dehydrogenase: 3616, glycoprotein: 1978, kinase: 9311, mitochondrial: 1965, polymerase: 3376, protease: 2234, receptor: 6952, reductase: 3746, synthase: 5246, sum: 40749
[Info] value_counts: full_protein_multimer: 115451, mul_protein_multimer: 6729, nan: 26, non_protein_monomer: 1547, non_protein_multimer: 2774, one_protein_multimer: 3856, protein_monomer: 73274, sum: 203657
[Info] value_counts: monomer: 73274, multimer: 115451, sum: 188725
[Info] 全部处理完成!
其他相关文章:
- PDB Database - RCSB PDB 数据集的多维度分析与整理
- PDB Database - AlphaFold DB PDB 数据集的多维度分析与整理
- PDB Database - ESM Atlas PDB 数据集的多维度分析与整理
参考:
- Applying Lambda functions to Pandas Dataframe
- Python | Convert a string representation of list into list