KDD2023 | Accelerating Antimicrobial Peptide Discovery with Latent Sequence-Structure Model

论文标题:Accelerating Antimicrobial Peptide Discovery with Latent Sequence-Structure Model
论文地址:https://dl.acm.org/doi/10.1145/3580305.3599249
代码:
GitHub - dqwang122/LSSAMP: Code for paper 'Accelerating Antimicrobial Peptide Discovery with Latent Sequence-Structure Model'
一、问题提出
典型的抗菌药物发现过程通常包括四个步骤:
基于现有AMP数据库构建候选库Candidate Library(应用人工启发式方法或深度生成模型来创建)
建立几个基于序列的过滤器来筛选候选肽不同的化学特征,包括计算指标以及理想属性。
将使用肽结构预测因子(如PEPFold 3)对这些序列的结构进行建模,并进行分子动力学模拟。
序列将在潮湿的实验室中合成和测试
近年来,深度生成模型使用序列属性来控制生成,直接生成具有理想属性的肽。然而,这些研究只考虑序列特征,而忽略了构效关系。生成的序列仍然需要输入到结构预测器中并手动检查,这减慢了发现过程。此外,该结构在确定生物属性方面也起着重要作用,从而促进了属性控制。
传统的AMP发现方法可分为三种方法: (i)模式识别算法。首先从现有AMP中构建抗菌序列模式数据库。每次选择模板肽用这些模式代替局部片段。(ii)遗传算法。使用AMP数据库设计一些抗菌活性功能,并利用这些功能优化祖先序列。(iii)分子建模和分子动力学方法。建立肽的3D模型,并通过肽与细菌膜之间的相互作用评估其抗菌活性
二、Method
Notations:肽是由氨基酸组成的短蛋白质,x = {x1; x2; · · · ; xL},位于第i位的氨基酸xi属于20种常见类型之一,也称为残基。二级结构描述肽的三维结构的局部形式。因此,肽结构表示为y= {y1;y2;···;yL},其中yi为第i位的二级结构标号,属于8种类型之一
深度生成模型通过将基于序列的过滤器与生成过程结合起来,直接创建具有用户指定属性的序列作为候选库,在加速AMP发现方面显示出了希望。然而,以往的研究只关注学习序列特征。
1、Antimicrobial Peptide Discovery
深度生成模型通过将基于序列的过滤器与生成过程结合起来,直接创建具有用户指定属性的序列作为候选库。然而,以往的研究只关注学习序列特征。在生成序列后,还需要通过外部工具对结构进行检查和过滤,使得生成过程效率低下。
使用对AMP机制至关重要的三个序列属性(电荷、疏水性、疏水力矩)来评估生成性能。通过限制α -螺旋结构,大多数结果都得到改进。结果表明,通过控制结构可以改善序列性能。因此,将结构信息纳入生成模型中,不仅可以将湿实验室前的所有步骤结合起来,加快发现速度,可以改善序列性质,提高生成过程效率。
2、VAE-based Generative Models
基于vae的多肽生成模型首先在无监督的肽或蛋白质序列上进行训练,然后在带有特定属性标签的特定序列上进行训练。
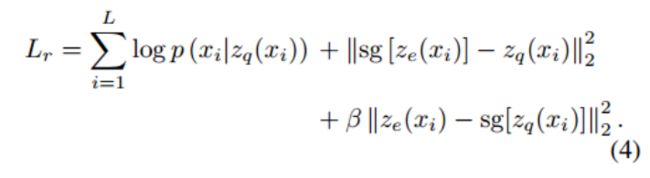
在潜在空间上增加一个8分类任务,以纳入二级结构信息。zq (xi)被送入一个单独的分类器进行结构预测,Ls与Lr类似,只是替换一部分损失:
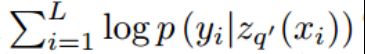
3、Multi-scale VQ-VAE
结构基序通常比序列模式长。例如,一个有效的α-螺旋至少包含4个残基,并且可能长于12个。具有特定生物学功能的序列模式要短得多,通常在1到8个残基之间。为捕获这些特征并将它们映射到相同的潜在空间,首先应用N个多尺度模式选择器Fn。然后,建立多个码本,查找嵌入zqn (xi)的最近的codebook,在序列重建和二级结构预测之间共享码本,以捕获残基与其结构之间的共同特征和关系。将串联的多尺度码本嵌入馈送到序列生成器:

基于方程4,多尺度VQ-VAE重构训练目标可以改编为多个码本上的损失之和:
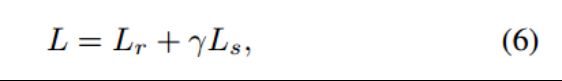
Training:
使用指数移动平均线(EMA)来更新码本中的embedding。保持一个count向量Ck,测量通过Eqn 3选择embedding向量ek作为ze(xi)的最近邻居的次数。Ck更新过程:
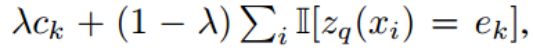
embedding ek更新过程:
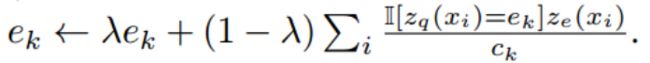
Prior Model
先验分布是一个分类分布,可以通过额外的先验模型实现自回归。为对z1:L之间的依赖关系进行建模,在code embedding上训练基于transformer的语言模型。从先前的模型中为每个码本n采样多个索引序列,然后查找codebook以得到embedding zqn。最后,将zqn输入到生成器和分类器中,生成具有二级结构的序列。还尝试通过现有的AMP结构模式来控制二次结构,以进一步提高生成质量。
三、Experiment
1、Experiment Setup
Dataset:下载蛋白质序列(550k),长度限制为100 (57k),Dr。然后使用AlphaFold社区版(ProSPr)预测二级结构。在过滤一些低质量的样本后,获得包含46k个样本的Ds,包括序列和二级结构信息。抗菌肽数据集,从抗菌肽数据库下载,筛选去重得到3222 AMPs,Damp
Dr、Ds随机抽取3000验证,3000测试。Damp,验证和测试的大小都是100
创建一组不含抗菌活性的负样本来比较。从Uniprot中去除具有抗菌活性的肽序列,以及长度< 10或> 40的序列,从而得到2021个非amp序列。
Implementation details :
encoder 、decoder基于2-layer Transformer,dim_model=128,head=8,dFFN=512,dropout=0.1,
classifier,使用CNN block,具有32个输入通道,膨胀尺度为[1;2;4;8;10]。 多尺度码本,将CNN作为F (n)提取特征。设n = 4,内核宽度范围为[1;2;4;8]。特征将被填充到与输入序列相同的长度。然后,使用K = 8和d = 128的4个码本。重构和预测具有相同的码本,即Nr = Ns = 4。提交系数设置为β = 0:05
Evaluation Metric:Uniqueness, Diversity, Similarity
唯一性是指在生成阶段中唯一肽的百分比。
多样性衡量的是所生成肽之间的相似性。计算每两个序列之间的Levenshtein距离,并通过序列长度对其进行归一化。然后对归一化距离求平均值,得到其多样性的均值。多样性越高,生成的多肽的差异性越大。
新颖性是生成肽和训练AMP之间的差异。对于每个生成的序列,在训练集中搜索与其Levenshtein距离最小的肽,并根据其长度对距离进行归一化。
2、Experimental Results
为每个baseline生成5000个序列
AMP Prediction:生成序列被预测为AMP的百分比。LSSAMP在7个分类器中的4个中表现最好,并且在所有分类器中具有最高的平均得分,表明其优于基线。peppcvae在AMPMIC和IAMPE预测器上表现最好,然而,它在其他预测器上表现不佳,平均得分较低。
Novelty:
Wet Laboratory Experiment:
选择了21种多肽,并测试了它们对三组革兰氏阴性菌(鲍曼氏杆菌、铜绿糖杆菌和大肠杆菌)的抗菌活性,耗时约30天。我们使用最小抑制浓度(MIC)来评估活性。LSSAMP可以有效地找到候选AMP,降低了时间成本。
Ablation Study:

Loss是验证集上的模型损失。AA Acc是一级的重构精度和SS Acc是二级结构的预测精度。第二阶段在大规模二级结构数据集Ds上训练模型将影响目标AMP数据集上的预测性能。如果删除多个子码本并使用相同大小的单个大码本,则性能会下降。
3、Analysis
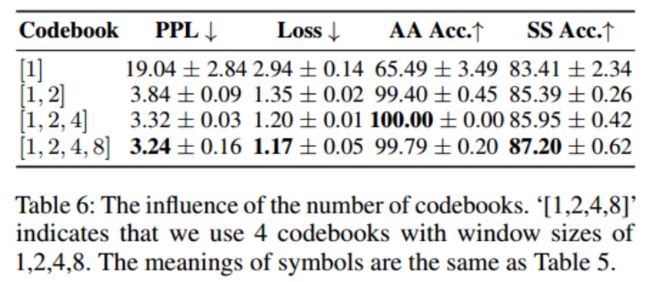
Codebook Number :不同数量的码本对生成性能的影响。从表6中,发现一个小的码本很难学习到足够的信息来重建序列。PPL, Loss和SS Acc随着密码本条目的增加而变得更好。当码本数为3时,重构精度达到最佳。这可能是由于序列的局部模式相对较短,window size=8太长:
Case Study :

在表7中展示了LSSAMP产生的10个肽,进一步利用PEPFold对生成的4个序列建立三维模型,并利用PyMOL绘制:

所有这些肽都有几个螺旋结构,这使它们更有可能具有抗菌能力。同时,虽然模型预测了ID = 2和ID = 3的长连续螺旋结构,但实际上在两个螺旋结构之间有一个小的线圈结构。这表明模型倾向于预测一个长连续的二级结构,而不是几个不连续的小碎片。
四、Conclusion
使用多尺度VQVAE对每个位置进行细粒度控制的LSSAMP。它将序列特征和结构特征映射到同一潜在空间中,通过对重叠分布进行采样,生成具有理想序列属性和二级结构的多肽。LSSAMP在AMP预测因子上表现良好,并设计了两种抗革兰氏阴性菌活性较高的肽。
1、Appendix
有几个物理性质对肽的抗菌活性至关重要。例如,带正电荷的氨基酸更容易与细菌膜结合,因为大多数细菌表面是阴离子,而那些具有高疏水性的氨基酸倾向于从溶液环境移动到细菌膜上。
Charge:肽序列S的总电荷定义为其所有残基C(xi)在pH 7.4时的电荷之和,即
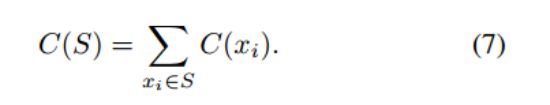
Hydrophobicity:疏水性反映了在细菌膜上结合脂质的倾向。具有高疏水性的肽很容易从溶液环境移动到细菌膜上。
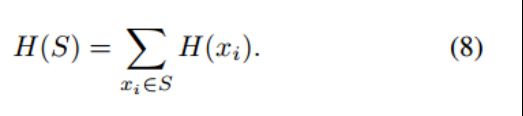
Amphipathicity / Hydrophobic Momentum:
amphipathicity(同时具有极性部分和非极性部分的分子) 同时结合水和脂质的能力,这是抗菌肽的明确特征。可以用疏水动量uH(S;θ)定义。疏水动量由每个残基xi的疏水性H(xi)以及残基之间的夹角θ决定:
该角度可由二级结构估算。α-螺旋结构θ为100◦,β-薄片结构θ为180◦。

2、Attribute Distribution
对于电荷,遵循专家总结的规则,选择净电荷为+2到+10的序列。如果APD的比例大于诱饵中的比例,我们将bin添加到评估度量的接受范围中。最后的范围是C2[2;10]、H2[0:25;1]和uH2[0:5;0:75][1:75;1]。
3、Secondary Structure Filter
AMP的生物学功能由其氨基酸序列和折叠结构决定。如果肽不能折叠成合适的结构,仍然很难发挥作用。例如,通过形成螺旋结构,肽可以在一侧聚集疏水性氨基酸和亲水性氨基酸。这种两亲性结构有助于肽插入膜中,并与膜中的其他分子保持稳定的孔:

蓝色表示疏水性氨基酸,红色表示亲水性氨基酸。在左边,尽管具有合理氨基酸的肽已经附着在细菌膜上,但它们仍然无法插入其中。然而,通过折叠成螺旋结构,如右边所示,肽保持了一个稳定的孔,该孔破坏了细菌的膜。
但控制二级结构是否也会影响序列属性?将生成的肽的二级结构控制为α-螺旋作为基线。性能差距:
通过将序列限制在α-螺旋结构中,大多数结果都得到了改进。结果表明,通过控制结构,可以提高序列属性,验证了在受控生成过程中引入二级结构的重要性。然而,序列大小已经显著减小,这表明这种先生成后过滤的管道效率低下。
4、Results of Sequence Attributes
使用上述三个序列属性来评估生成性能。
Structure Condition
控制二级结构可以影响生成的肽的属性。因此,限制具有不同比例的coil结构的百分比:
随着螺旋结构数量的减少,阳性肽的百分比不断增加。然而,对于疏水性和疏水力矩,百分比在0.3之后下降。因此,在主要实验中,将螺旋结构的长度限制在30%。
Visualization of Residue Distribution
为了说明生成的肽中残基的分布,绘制了tSNE图。变换向量,每个维度代表某个残基代表肽的概率。然后使用tSNE将高维向量转换为2D并将其可视化。发现LSSAMP w/o条件和APD之间有很大的重叠,这表明模型已经捕捉到了APD的全局分布:
Visualization of LSSAMP Distribution
绘制了APD、Decoy和我们的模型的残基、电荷、序列长度、疏水性和疏水动量的分布。在没有条件的情况下,LSSAMP的分布与APD相似,这表明LSSAMP成功地学习了AMP的序列分布。