Rosetta Tutorial 11 & 12 翻译 | 主要为 Relax 部分
该文档翻译自Rosetta官网的教程性文档 Rosetta Tutorials, Demos, and Protocol Captures,原文链接:https://www.rosettacommons.org/demos/latest/Home 。“”内的内容为译者添加。
Contents
- 11. Relax: Refining Structures
-
- 11.1. The Relax Protocol
- 11.2. Basic Demo
- 11.3. Modifying the scope of Relax
-
- 11.3.1. Restricting the conformations that Relax can sample
-
- 11.3.1.1 By MoveMap
- 11.3.1.2 By Completely Disallowing Flags
- 11.3.1.3 By Reference Structure
- 11.3.3. Restricting the sequence it can sample
- 12. Constraints: Biasing Towards a Structure
-
- 12.1. Introduction
- 12.2. Demo
- 12.3. More Demos
11. Relax: Refining Structures
11.1. The Relax Protocol
弛豫(relax)是 Rosetta 中放松结构的主要 protocol;也就是说,它在3D空间中对接近给定结构的构像进行采样,以找到评分最低的变体,然后交替运行封装器和最小化器。这通常是为了允许在不同的结构(包括晶体结构和 Rosetta 的采样 protocols 的输出)之间进行同类比较(apples-to-apples comparison 哈哈),当然首先根据相同的分数函数在局部空间中最小化它们。因此,明智的做法是在任何你打算相互比较的结构上运行弛豫。
11.2. Basic Demo
进入score_jd2打分命令,将会得到一个score_crystal.sc文件,后面有用;运行第三行带有标签文件general_relax_flags的relax命令,
cd <path_to_Rosetta_directory>/demos/tutorials/Relax_Tutorial
$ROSETTA/main/source/bin/score_jd2.default.linuxclangrelease -s 1ubq.pdb -out:suffix _crystal @crystal_score_flags
$ROSETTA/main/source/bin/relax.default.linuxclangrelease -s 1ubq.pdb -out:suffix _relaxed @general_relax_flags
标签文件general_relax_flags内容如下:
-nstruct 2 # 在输入结构上运行两个独立的弛豫轨迹
-relax:default_repeats 5 # 运行以侧链重新封装和最小化 5 个周期的弛豫算法
-out:path:pdb ./tutorial_output # 将弛豫结构输出到 ./tutorial_output/
-out:path:score ./expected_output # 将打分文件输出到 ./expected_output/
我们将会在tutorial_output/文件夹中生成两个结构1ubq_relaxed_0001.pdb和1ubq_relaxed_0002.pdb,在expected_output/文件夹中生成score_relax.sc。打开这个分数文件,可以发现弛豫前后结构差异相对较小,但得分的显著差异,这可能是 300-500 REU,该值取决于放松的成功。在第一种情况下,1ubq.pdb 直接从蛋白质数据库中提取,并根据晶体学能量函数进行优化;在第二种情况下,Rosetta 根据蛋白质的内部评分功能将蛋白质移动到最优状态 没弄明白这里的case是什么意思,也不知道咋突然冒出来这两句。
score_relax.sc内容及结构对比结果:
SEQUENCE:
SCORE: total_score dslf_fa13 fa_atr fa_dun fa_elec fa_intra_rep fa_intra_sol_xover4 fa_rep fa_sol hbond_bb_sc hbond_lr_bb hbond_sc hbond_sr_bb lk_ball_wtd omega p_aa_pp pro_close rama_prepro ref yhh_planarity description
SCORE: 32.678 0.000 -397.647 288.599 -113.091 1.777 16.826 57.039 242.952 -7.389 -23.132 -1.549 -18.828 -8.756 1.713 -12.808 2.383 -7.297 11.884 0.000 1ubq_crystal_0001
SCORE: -255.900 0.000 -422.146 82.390 -138.558 0.814 12.643 46.385 255.134 -14.981 -24.237 -10.404 -21.808 -10.916 5.207 -16.226 0.107 -11.210 11.884 0.022 1ubq_relaxed_0001
SCORE: -258.089 0.000 -418.894 85.650 -149.335 0.813 13.055 45.762 257.470 -13.980 -24.088 -12.497 -22.465 -8.380 3.995 -15.731 0.161 -11.548 11.884 0.037 1ubq_relaxed_0002
Executive: RMSD = 0.328 (65 to 65 atoms)

为了进一步研究这一点,在以下命令中手动将“N”更改为1到10之间的整数;然后,绘制循环数与平均得分之间的关系放了一个 shell 脚本在下面。
$ROSETTA/main/source/bin/relax.default.linuxclangrelease -s 1ubq.pdb -out:suffix _N_relax_cycles -relax:default_repeats N -nstruct 10 -out:path:pdb ./tutorial_output -out:path:score ./expected_output
# script
for(( var=1 ; $var <= 10 ; var++ ))
do
$ROSETTA_BIN/relax.default.linuxgccrelease -s 1ubq.pdb -out:suffix _${var}_relax_cycles -relax:default_repeats $var -nstruct 10 -out:path:pdb ./tutorial_output -out:path:score ./expected_output
done

您应该会看到值的递减,特别是对于非常大的N,以及从初始结构逐渐增加的发散。最好使用 5 到 15 个弛豫周期,通常 5 个就足够了。5 为默认值,因此-relax:default_repeats选项通常可以省略 确实如此。
11.3. Modifying the scope of Relax
11.3.1. Restricting the conformations that Relax can sample
11.3.1.1 By MoveMap
默认情况下,允许弛豫选择新的侧链 rotamers、移动蛋白质主链或移动蛋白质亚基相对位置。虽然这允许 protocol 找到更优的解决方案,但限制弛豫以防止以与生物数据背道而驰的方式修改结构可能有帮助,例如在确保所设计的蛋白与其天然配体之间不会发生相对运动尤为有用。你可以通过使用选项-in:file:movemap为弛豫步骤提供 MoveMap。运行下列语句来康康MoveMap会对弛豫造成怎样的影响:
$ROSETTA/main/source/bin/relax.default.linuxclangrelease -s 1ubq.pdb @general_relax_flags -out:suffix _lever_arm -in:file:movemap lever_arm_movemap
lever_arm_movemap内容:
RESIDUE * NO
RESIDUE 1 10 BBCHI
输出文件score_lever_arm.sc内容:
SEQUENCE:
SCORE: total_score dslf_fa13 fa_atr fa_dun fa_elec fa_intra_rep fa_intra_sol_xover4 fa_rep fa_sol hbond_bb_sc hbond_lr_bb hbond_sc hbond_sr_bb lk_ball_wtd omega p_aa_pp pro_close rama_prepro ref yhh_planarity description
SCORE: 32.678 0.000 -397.647 288.599 -113.091 1.777 16.826 57.039 242.952 -7.389 -23.132 -1.549 -18.828 -8.756 1.713 -12.808 2.383 -7.297 11.884 0.000 1ubq_crystal_0001
SCORE: -185.516 0.000 -386.063 85.558 -132.678 0.808 13.697 61.265 242.113 -7.022 -23.341 -7.977 -18.632 -7.028 1.824 -13.272 0.593 -7.245 11.884 0.000 1ubq_lever_arm_0001
SCORE: -186.282 0.000 -388.269 86.566 -131.753 0.803 13.707 61.012 240.748 -6.942 -23.339 -6.868 -18.195 -7.004 1.615 -13.485 0.593 -7.355 11.884 0.000 1ubq_lever_arm_0002
结构对比:
![]()

观察到所有可以移动的C端的骨架都移动了。这是使用内部坐标的移动地图所特有的,被称为杠杆臂效应;虽然 Rosetta 中的一些 protocols 是考虑到这一点而编写的,但如果没有在其movemap中声明禁止的话,弛豫会允许杠杆臂效果,
11.3.1.2 By Completely Disallowing Flags
除了MoveMap,你可以在命令行中使用标签:
-relax:chi_move false
-relax:bb_move false
-relax:jump_move false
来完全禁止侧链、骨架及不同结构之间的相对运动;运行下述语句来康康这些标签怎么发挥作用的:
$ROSETTA/main/source/bin/relax.default.linuxclangrelease -s 1ubq.pdb -relax:bb_move false -out:suffix _no_sidechain_motion @general_relax_flags
输出文件score_no_sidechain_motion.sc内容:
SCORE: total_score dslf_fa13 fa_atr fa_dun fa_elec fa_intra_rep fa_intra_sol_xover4 fa_rep fa_sol hbond_bb_sc hbond_lr_bb hbond_sc hbond_sr_bb lk_ball_wtd omega p_aa_pp pro_close rama_prepro ref yhh_planarity description
SCORE: 32.678 0.000 -397.647 288.599 -113.091 1.777 16.826 57.039 242.952 -7.389 -23.132 -1.549 -18.828 -8.756 1.713 -12.808 2.383 -7.297 11.884 0.000 1ubq_crystal_0001
SCORE: -193.575 0.000 -382.042 92.992 -134.860 0.832 13.814 44.901 243.360 -6.990 -23.132 -10.407 -18.828 -6.797 1.713 -12.808 0.086 -7.297 11.884 0.005 1ubq_no_sidechain_motion_0001
SCORE: -193.576 0.000 -382.061 93.021 -134.930 0.831 13.808 44.905 243.381 -6.985 -23.132 -10.396 -18.828 -6.776 1.713 -12.808 0.085 -7.297 11.884 0.006 1ubq_no_sidechain_motion_0002
并将tutorial_output/中的一个或两个输出结构对齐到原始的1ubq.pdb,你应该看到在弛豫之前(蓝色)和之后(玫红色),你的骨架几乎没有变化,但同时弛豫后具有较高的最终能量。
11.3.1.3 By Reference Structure
你也可以不通过完全禁止的标签阻止弛豫中的剧烈动作,这可以通过添加约束来实现:
-relax:constrain_relax_to_start_coords
-relax:constrain_relax_to_native_coords -in:file:native <filename.pdb>
第一行阻止与初始输入结构不同的输出;第二行阻止与额外提供的输入文件差异较大的输出。顾名思义,这对于确保对某种原生结构的忠实性特别有用。这些被执行为调和约束,所以线性的发散反映在分数的二次增长中。可以修改通过设置-relax:coord_cst_width ,这将超过一定距离 的正态谐波约束变为平坦的谐波约束,如此任何与输入文件相差 之内的输出变化将不会被罚分,从而允许小的变化。运行下述语句以尝试(general_relax_flags内容在 11.2. 已介绍过):
$ROSETTA/main/source/bin/relax.default.linuxclangrelease -s 1ubq.pdb -out:suffix _constrained_relax -relax:constrain_relax_to_start_coords @general_relax_flags
score_constrained_relax.sc内容及结构对比结果:
SCORE: total_score dslf_fa13 fa_atr fa_dun fa_elec fa_intra_rep fa_intra_sol_xover4 fa_rep fa_sol hbond_bb_sc hbond_lr_bb hbond_sc hbond_sr_bb lk_ball_wtd omega p_aa_pp pro_close rama_prepro ref yhh_planarity description
SCORE: 32.678 0.000 -397.647 288.599 -113.091 1.777 16.826 57.039 242.952 -7.389 -23.132 -1.549 -18.828 -8.756 1.713 -12.808 2.383 -7.297 11.884 0.000 1ubq_crystal_0001
SCORE: -257.968 0.000 -410.742 80.231 -135.018 0.791 12.561 45.034 238.217 -12.724 -24.337 -9.506 -21.996 -9.437 5.887 -16.729 0.096 -12.186 11.884 0.004 1ubq_constrained_relax_0001
SCORE: -258.478 0.000 -413.538 82.930 -139.068 0.791 12.492 45.171 245.225 -8.765 -24.277 -13.107 -22.011 -9.055 4.081 -17.781 0.121 -13.593 11.884 0.020 1ubq_constrained_relax_0002
Executive: RMSD = 0.355 (68 to 68 atoms)

可以发现与晶体结构(蓝色)、11.2.不加约束的弛豫结构(绿色)相比,添加参考约束的弛豫结构(黄色)变化介于前两者之间
11.3.2. Changing the behavior of constraints
默认情况下,随着运行的进行,弛豫会逐渐减少给定约束的权重,以便在优化最终结构的同时探索更多的约束空间。如果必须时刻维持约束,那么上述行为可以用-relax:ramp_constraints false标签关闭。当使用-relax: constrain_relax_to_start_coordinates和类似的选项时,这个标签特别有用,因为它使输出坐标比倾斜约束运行更接近指定坐标。
11.3.3. Restricting the sequence it can sample
默认情况下,弛豫不会改变输入序列。你可以通过给出一个 resfile 并使用标签-relax:respect_resfile -packing:resfile *resfile*来允许这一操作。它将设置其内部封装器以遵从所提供的 resfile。但这只控制了包装行为,没有改变最小化步骤;它也可以用来增加关键残基周围的 rotamer 采样,但它本身不能确保特定的 rotamers 被保存。你还可以通过使用-disable_design false选项来允许弛豫全局更改序列。
下面是一个演示,请运行:
$ROSETTA/main/source/bin/relax.default.linuxclangrelease -s 1ubq.pdb -out:suffix _relaxed_with_resfile -relax:respect_resfile -packing:resfile 1ubq.resfile @general_relax_flags
# 1ubq.resfile 内容;不过NATRO是不改变序列,
NATRO
输出文件score_relaxed_with_resfile.sc内容:
SCORE: total_score dslf_fa13 fa_atr fa_dun fa_elec fa_intra_rep fa_intra_sol_xover4 fa_rep fa_sol hbond_bb_sc hbond_lr_bb hbond_sc hbond_sr_bb lk_ball_wtd omega p_aa_pp pro_close rama_prepro ref yhh_planarity description
SCORE: 32.678 0.000 -397.647 288.599 -113.091 1.777 16.826 57.039 242.952 -7.389 -23.132 -1.549 -18.828 -8.756 1.713 -12.808 2.383 -7.297 11.884 0.000 1ubq_crystal_0001
SCORE: -211.918 0.000 -407.523 119.760 -134.079 0.924 16.740 44.392 237.633 -14.237 -25.817 -9.014 -20.916 -10.109 3.134 -16.834 0.199 -8.058 11.884 0.001 1ubq_relaxed_with_resfile_0001
SCORE: -211.918 0.000 -407.523 119.760 -134.079 0.924 16.740 44.392 237.633 -14.237 -25.817 -9.014 -20.916 -10.109 3.134 -16.834 0.199 -8.058 11.884 0.001 1ubq_relaxed_with_resfile_0002
Executive: RMSD = 0.569 (72 to 72 atoms)
结构对比显示带有 resfile 的弛豫结构变化小于 11.2. 的弛豫结构 不知道为啥。
我们不妨大胆一些,把1ubq.resfile的内容换成ALLAA(不过这样尝试的 rotamer 会贼多,所以耗时也会比较久),然后运行:
$ROSETTA_BIN/relax.default.linuxgccrelease -s 1ubq.pdb -out:suffix _relaxed_with_newresfile -relax:respect_resfile -packing:resfile 1ubq.resfile @general_relax_flags
输出文件score_relaxed_with_newresfile.sc内容:
SEQUENCE:
SCORE: total_score dslf_fa13 fa_atr fa_dun fa_elec fa_intra_rep fa_intra_sol_xover4 fa_rep fa_sol hbond_bb_sc hbond_lr_bb hbond_sc hbond_sr_bb lk_ball_wtd omega p_aa_pp pro_close rama_prepro ref yhh_planarity description
SCORE: 32.678 0.000 -397.647 288.599 -113.091 1.777 16.826 57.039 242.952 -7.389 -23.132 -1.549 -18.828 -8.756 1.713 -12.808 2.383 -7.297 11.884 0.000 1ubq_crystal_0001
SCORE: -307.973 0.000 -439.807 78.061 -143.168 1.073 12.444 57.989 203.384 -12.554 -24.521 -7.478 -19.444 -8.467 4.195 -30.343 0.605 -16.566 36.317 0.306 1ubq_relaxed_with_newresfile_0001
SCORE: -311.881 0.000 -461.886 84.110 -128.158 0.959 12.023 62.479 210.149 -17.084 -24.917 -4.720 -21.250 -12.106 4.587 -27.744 0.919 -16.263 26.760 0.261 1ubq_relaxed_with_newresfile_0002
序列变化:
![]()
结构比对(蓝色为晶体结构,橙色为NATRO设置的弛豫结构,紫色为ALLAA设置的弛豫结构):
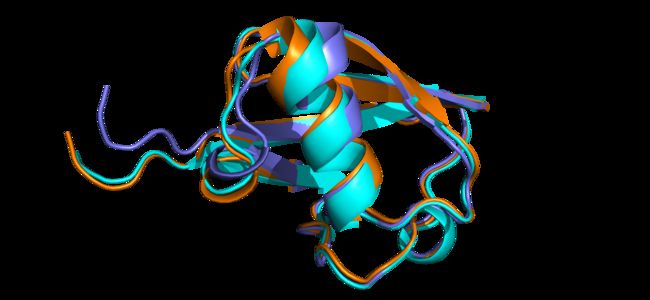
12. Constraints: Biasing Towards a Structure
KEYWORDS: DESIGN GENERAL
Tutorial by Frank David Teets ([email protected]). Edited by Sebastian Rämisch ([email protected]) and Kristin Blacklock ([email protected]). File created 21 June 2016 as part of the 2016 Documentation eXtreme Rosetta Workshop (XRW).
12.1. Introduction
用户希望使用 Rosetta 解决的许多生物学问题涉及一些具体的生物学或功能方面的考虑,这些可能无法在 PDB 文件中反映或通过正常的评分函数进行评估。约束(constraints)是衡量一个结构是否符合这些额外考虑的一般方法;例如,可能希望在弛豫时使用约束,以确保维持可能的二硫键。约束的工作方式如下:
-
对给定的构象进行某些计算(例如其中某个键的键长为3.2 Å);
-
有一个合适的评估函数来描述哪些值是好的,哪些值是坏的;例如,理想键长可以是二次函数(调和势)的最小值。然后通过评估函数对测量值进行罚分;对于调和势,当测量的键长距离理想键长越远,罚分越大。
-
这个罚分乘以一个权重因子,并入到总能量中。
例如,可以设置一个简单的约束来测量两个原子之间的距离,减去理想的距离,然后从分数中减去这个差。这个约束是这样的:
AtomPair CA 20 CA 6 LINEAR_PENALTY 9.0 0 0 1.0
该约束定义由两部分组成。AtomPair CA 20 CA 6表示要测量什么,而LINEAR_PENALTY 9.0 0 0 1.0 1.0定义如何将测量转换为罚分。
约束从几何属性的定义开始。在这种情况下,因为我们想要约束两个原子彼此之间的距离,所以我们需要一个AtomPair约束。接下来的四个字段是AtomPair约束的参数。具体来说,我们将两个原子定义为残基 20 和 6 的α-碳原子(Rosetta 编号)。以下是一些常用约束选项:
AtomPair和AtomAngle约束使用如上所述的参数化,而AtomAngle约束需要三个原子(顶点原子列在第二)。对侧链原子的AtomPair和AtomAngle约束对残基种类的变化没有鲁棒性。约束是根据起始结构中的原子本身建立的。当残基改变时,这种对应可能会变得不准确。此外还有NamedAtomPair和NamedAtomAngle约束,它们将始终约束命名原子,即使当残基种类发生变化时。AtomPair / NamedAtomPair值以 Å 为单位,而AtomAngle / NamedAtomAngle结果以弧度形式返回。
CoordinateConstraints的工作原理类似于AtomPair约束,只是第二个“原子”是3D空间中的一个点,而不是一个实际的原子。因为 Rosetta 使用了相对坐标,所以CoordinateConstraints需要第二个原子来定义坐标轴;第二个原子不应该与第一个原子协同运动,但也不需要与第一个原子有任何特定的距离。相似的,LocalCoordinateConstraint使用额外三个原子来定义坐标框架。这些可以通过constrain_to_native_coord在弛豫运行中直接使用。
在约束的参数之后是评分函数的定义,它将测量转化为能量罚分。这里我们使用的是LINEAR_PENALTY函数。函数选择之后的参数提供了关于函数形式的详细信息。LINEAR_PENALTY的第一个参数表示最低能量在原子相距 9.0 Å时。根据约束条件,如果输入结构是“完美的”,那么残基 6 和残基 20 的α -碳原子之间将相隔 9 Å。剩下的字段定义在与 9.0 Å 理想值不同时,分数的变化情况。对于LINEAR_PENALTY,分数随着理想值的(绝对)差线性增加。斜率可以通过LINEAR_PENALTY的第四个参数来控制(这里是1.0)。LINEAR_PENALTY还包括添加一个平坦区域的功能,函数在其中返回一个常量值。这里,这个值是 0,平坦区域的宽度也是 0。
其他常见的评分函数有:HARMONIC是理想值和实际值之间的距离的平方,通常用于各种类型的距离约束。CIRCULARHARMONIC是角当量。
除了上述例子给出了距离约束方式外,还有:扭转约束、其他角度限制、模棱两可的约束(Ambiguous constraints)、密度约束等。要了解有关 Rosetta 可以施加的其他约束类型和函数类型的更多信息,请参阅约束文件文档。
12.2. Demo
为了验证上述介绍,首先通过以下命令对泛素进行弛豫:
cd $ROSETTA3/demos/tutorials/Constraints_Tutorial/
# 无约束
$ROSETTA3/bin/relax.default.linuxclangrelease -s 1ubq.pdb -out:suffix _unconstrained @general_relax_flags
# 有约束
$ROSETTA3/bin/relax.default.linuxclangrelease -s 1ubq.pdb @general_relax_flags -out:suffix _unreasonably_constrained @unreasonable_constraint_flags
# unreasonable_constraint_flags 内容
-constraints:cst_fa_file unreasonably_constrained_atompairs.cst
-constraints:cst_fa_weight 100
# unreasonably_constrained_atompairs.cst 内容
AtomPair CA 5 CA 25 HARMONIC 1 1 10
# 无约束
SCORE: total_score dslf_fa13 fa_atr fa_dun fa_elec fa_intra_rep fa_intra_sol_xover4 fa_rep fa_sol hbond_bb_sc hbond_lr_bb hbond_sc hbond_sr_bb lk_ball_wtd omega p_aa_pp pro_close rama_prepro ref yhh_planarity description
SCORE: -258.788 0.000 -418.767 83.861 -149.348 0.807 14.074 47.799 255.711 -15.137 -23.846 -13.809 -22.166 -8.461 6.003 -16.513 0.112 -10.997 11.884 0.003 1ubq_unconstrained_0001
# 有约束
SCORE: total_score angle_constraint atom_pair_constraint coordinate_constraint dihedral_constraint dslf_fa13 fa_atr fa_dun fa_elec fa_intra_rep fa_intra_sol_xover4 fa_rep fa_sol hbond_bb_sc hbond_lr_bb hbond_sc hbond_sr_bb lk_ball_wtd omega p_aa_pp pro_close rama_prepro ref yhh_planarity description
SCORE: 253.633 0.000 229.692 0.000 0.000 0.000 -324.135 89.575 -87.506 1.080 12.616 172.099 218.882 -16.360 -4.337 -6.576 -16.926 -14.086 8.325 -15.374 0.671 -5.895 11.884 0.006 1ubq_unreasonably_constrained_0001
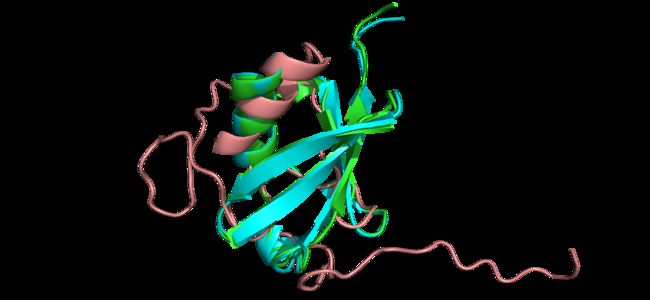
原生 PDB 1UBQ(蓝色),无约束的精化版本(绿色)和不合理的约束精化版本(鲑鱼色)。
在unreasonable_constraint_flags文件中,尝试使用-constraint:cst_fa_weight标志改变约束的权重,并分析这如何改变最终的结构。高权重(>1000)应该产生明显的物理结构,除非你将理想约束设置为接近其原生值的东西。重要的是选择一个与预期得分成比例的权重,以及你希望你的结果如何符合约束;一次 预弛豫 运行应该会让你对预期的分数范围有一个概念。同样重要的是,所选择的约束函数反映了生物系统的任何不确定性,因此存在一些约束函数的“平坦”区域(其中包括LINEAR_PENALTY和HARMONIC),这些约束函数允许在一个范围内的任何值等价计算。
12.3. More Demos
如上所述,约束是使 Rosetta 的分数反映有关被评分(或设计)系统的一些实验数据的一种方法,并且不支持与数据冲突的结构。举个例子,假设我们希望弛豫蛋白质-蛋白质相互作用的一半,但也许我们从突变研究中知道,每个亚基上存在某些残基的相互作用。通过约束包含这些相互作用信息可能是有意义的,像4eq1.pdb中的A链 G356-B链 L423、A链 L423-B链 G356这样。所以我们可以特定的约束这几个位置 原文写得是AtomPair CA 432A CA 356B,但安装包的constrained_atompairs.cst中写得是AtomPair CA 423A CA 356B,事实上也应该是423A,并运行:
# constrained_atompairs.cst 内容
AtomPair CA 356A CA 423B HARMONIC 4.3 0.25 1
AtomPair CA 423A CA 356B HARMONIC 4.3 0.25 1
# constraint_flags 内容
-constraints:cst_fa_file constrained_atompairs.cst
-constraints:cst_fa_weight 10
# 无约束
$ROSETTA3/bin/relax.default.linuxclangrelease -s 4eq1.pdb -out:suffix _unconstrained -ignore_unrecognized_res @general_relax_flags
# 有约束
$ROSETTA3/bin/relax.default.linuxclangrelease -s 4eq1.pdb -out:suffix _constrained -ignore_unrecognized_res @general_relax_flags @constraint_flags
# 无约束
SCORE: total_score dslf_fa13 fa_atr fa_dun fa_elec fa_intra_rep fa_intra_sol_xover4 fa_rep fa_sol hbond_bb_sc hbond_lr_bb hbond_sc hbond_sr_bb lk_ball_wtd omega p_aa_pp pro_close rama_prepro ref yhh_planarity description
SCORE: -734.356 0.000 -1380.547 289.929 -422.259 2.619 48.342 154.097 828.312 -58.158 -89.724 -48.103 -53.192 -28.665 21.481 -41.312 0.532 -28.356 70.537 0.111 4eq1_unconstrained_0001
# 有约束
SCORE: total_score angle_constraint atom_pair_constraint coordinate_constraint dihedral_constraint dslf_fa13 fa_atr fa_dun fa_elec fa_intra_rep fa_intra_sol_xover4 fa_rep fa_sol hbond_bb_sc hbond_lr_bb hbond_sc hbond_sr_bb lk_ball_wtd omega p_aa_pp pro_close rama_prepro ref yhh_planarity description
SCORE: -731.853 0.000 1.336 0.000 0.000 0.000 -1369.581 288.447 -413.002 2.624 45.977 151.931 817.137 -62.383 -90.951 -46.341 -53.211 -26.648 17.087 -37.634 0.593 -27.848 70.537 0.075 4eq1_constrained_0001
# 只有第一个约束
SCORE: total_score angle_constraint atom_pair_constraint coordinate_constraint dihedral_constraint dslf_fa13 fa_atr fa_dun fa_elec fa_intra_rep fa_intra_sol_xover4 fa_rep fa_sol hbond_bb_sc hbond_lr_bb hbond_sc hbond_sr_bb lk_ball_wtd omega p_aa_pp pro_close rama_prepro ref yhh_planarity description
SCORE: -720.681 0.000 0.562 0.000 0.000 0.000 -1363.733 287.406 -408.967 2.622 46.618 155.946 812.203 -60.476 -90.284 -47.742 -52.670 -25.896 20.431 -40.044 0.601 -27.875 70.537 0.082 4eq1_single_constrained_0001
您应该看到,当每个亚单位的其余部分移动时,每个亚单位的N端相对于残基423的移动非常小,这与我们输入的约束条件一致(您可以通过测量另一个亚基上的残基356到残基423的距离来更严格地验证这一点量了一下没发现有啥区别,在关闭第二个限制之后依然没啥区别,不清楚为啥,可能对这个蛋白来说现在的算法已经好到不需要加约束了)。正如前面在弛豫教程中提到的,如果我们希望防止任何氨基酸从起始位置移动得特别远,我们可以使用该选项-relax:constrain_to_starting_coords 不理解咋突然蹦出来这句话。