图神经网络GNN GCN AlphaFold2 虚拟药物筛选和新药设计
文章目录
- 图神经网络
- 1. Geometric Deep Learning
-
- Representation learning 表征学习
- 机器学习的数据类型:序列、网格、图
- 引出GNN
- 2. Graph Neural Networks
-
- Machine Learning Lifecycle
- learning graph is hard
- Feature Learning in Graphs
- Ways to Analyze Networks
- A Naive Approach 介绍一种最初级的学习图的方式
- 一种更好的处理方式:深度图编码器(Deep Graph Encoders)
- Graph Convolutional Networks(GCN)
-
- Idea: Aggregate Neighbors
- The three “flavours" of GNN layers
- 3. Deep Learning on Graphs
-
- Deep Encoder
- GraphSAGE
- Application to Drug Design/Evaluation
- Heterogenous Networks
- Training the Model
- 4. GNNs for Protein folding
-
- Chemical Structures as Graphs
- Protein Structure Prediction
-
- Methods for Protein Structure Prediction
- Old method: fragment assembly
- New Strategy
-
- Co-evolution Analysis
- Towards An End-to-End Workflow
- AlphaFold2 architecture
-
- 补充:跟李沐精读论文
- 5. Computational Drug Development
-
- Computational drug discovery: three schemes
- 6. Virtual drug screening
-
- Virtual screening
- Part 1: antibiotic discovery 抗生素
-
- Traditional approach: hand-crafted features
- Graph neural network (GNN)
- Use GNN for virtual screening
- 7. De novo drug design
-
- Previous solution 1: sequence-based methods
- Junction tree variational autoencoder
- Details: hierarchical encoder & decoder
- Motif-by-motif versus node-by-node
- 8. Research Frontiers
来自Manolis Kellis教授(MIT计算生物学主任)的课《人工智能与机器学习》,中间结合李沐老师的两个精读视频(GNN和AlphaFold2)作为补充
本节课主要介绍了几何深度学习、图神经网络
主要内容有图神经网络、GNN、GCN、对称性、等变性、信息传递、蛋白质空间结构预测(AlphaFold2)、药物设计(虚拟药物筛选和新药设计)
本节课的内容比较多,按照目录/大纲点击跳转到需要的模块就行
有需要课程视频的可以私信发油管链接
AI for Drug Design - Lecture 16 - Deep Learning in the Life Sciences (Spring 2021)
Deep Learning for Protein Folding - Lecture 17 - MIT Deep Learning in Life Sciences (Spring 2021)
图神经网络
快速预习三件套(1h):
李沐老师论文精读视频:零基础多图详解图神经网络(GNN/GCN)【论文精读】
博客文章链接:A Gentle Introduction to Graph Neural Networks
博客的博客(方便回顾):【李沐精读GNN论文总结】A Gentle Introduction to Graph Neural Networks
进阶、更全面(3h):
唐宇迪AI:【唐博士带你学AI】2022最新图神经网络课程
当然了,图神经网络的内容远远不是几个小时可以学完的,以上的内容都仅仅算是入门。GNN存在一定的门槛,要先有一定的深度学习基础。
刚复习完一些图论就来学图神经网络里,感受到了图论和机器学习的结合,好玩捏
本次是第六周的课程,讲座主要内容如下:
本讲座涵盖了几何深度学习和图神经网络。几何深度学习是一种从图中学习的框架。图神经网络是一种可以从图表示学习的深度学习网络,可以用于处理大语言模型的顺序数据,蛋白质折叠,药物设计和计算药物开发。讲师介绍了对称性、变化和消息方式的概念。
Manolis 正在讨论使用网络和图来表示复杂的3D网络。他解释说网络可以被表示为图,这可以用来表示用户和对象之间的关系。他举了一个例子,说明化学网络是通过化学键的图表来表示的,大脑是通过神经元网络来表示的。Manolis正在寻找一种原始的方法来用邻居表示生物网络。当前的方法使用卷积神经网络,其中每个节点都连接到所有邻居,但这有太多的参数,不能适用于不同大小的图。Manolis 建议使用图神经网络,其中节点连接到他们的邻居,然后根据每个节点的邻居学习特征。
Manolis 讨论了图神经网络如何应用于计算药物开发,虚拟药物筛选和新药设计。他解释说,化学结构可以被表示为图,这与化学图表是一样的。他展示了如何比较这些图来创建一个化学指纹。
Manolis 在图上进行了智慧发现柠檬测试。该测试问的是,每个节点可以根据其邻居计算什么功能。Manolis查看每个节点的属性,并根据每个节点的邻居进行命名。这表明在图中有两种类型的节点:有3个邻居的节点和有2个邻居的节点。
Manolis想知道我们是否能通过在图中编码蛋白质来预测他们的性质,比如他们在三维空间中的结构,从而了解化学、化学和生物学的一些信息。Manolis解释说,蛋白质的结构是由其氨基酸序列决定的。有许多蛋白质结构预测的方法,包括模板基础建模,该方法使用一个模板作为模板来模拟任何预测中的蛋白质。最成功的方法是多序列比对,该方法将序列对齐到已知的片段,并模拟他们如何配合。
Manolis进行了计算药物发现的演示。主要有三种方法:1.虚拟筛选:筛选大量化合物并测试它们是否符合目标。2.模拟:模拟化学空间并预测它在三维中如何折叠。3.新药设计:预测细菌对抗生素的抗性。
Manolis展示了如何使用层次化的编码器和解码器在一个交叉树中表示一个分子图。目标是从分子图的简化表示中生成有意义的分子。Manolis展示了如何使用一个基于图神经网络的动机网络对分子进行编码和解码。
Manolis对他和他的团队在深度学习和图神经网络方面所做的研究进行了概述。他们研究了如何通过学习邻居的属性并将它们传递给节点本身来建模图中节点之间的关系。这使他们能够根据邻居学习更多的属性,然后通过网络的不同层次传播这些属性,并对邻居层的嵌入进行平均。这项研究已经导致了两种类型的应用:筛选分子的属性并根据这些属性生成新的药物。
- 封面(封面下半部分做错了hh)

- 本节课大纲

1. Geometric Deep Learning
Representation learning 表征学习
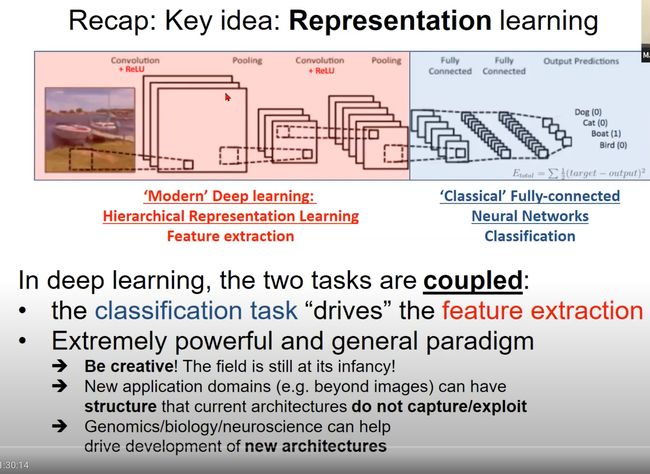
- 传统的深度学习核心是一种表征学习,一般由两部分组成
- ‘Modern’ Deep learning:现代深度学习模型,如卷积神经网络(CNN)和循环神经网络(RNN)。是一种层次化表征学习,在深度学习中,从底层特征到高层特征的学习过程被视为层次化的表示学习。
- ‘Classical’ Fully-connected Neural Networks:传统的全连接神经网络,用于分类
- 在深度学习中,特征提取和分类任务是相互关联(耦合)的:分类任务“驱动”特征提取
- 深度学习是一个非常强大且通用的范例。在深度学习的领域,人们需要具有创造性,因为该领域仍处于其发展的初期阶段。新的应用领域(例如,超越图像的领域)可能具有当前架构无法捕捉/利用的结构。基因组学/生物学/神经科学可以帮助驱动新架构的开发。
机器学习的数据类型:序列、网格、图

- 我们目前学习到的CNN和RNN之类的,都是处理规则的、结构化的网格数据。
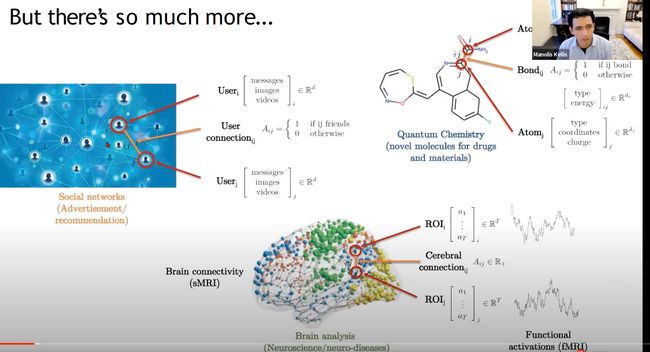
- 但是世界上还存在更多的,非网格数据,图数据。比如社交网络(搜广推常用)、化学分子、人脑神经元等等
- 要如何表示和学习这些数据呢,有人说用邻接矩阵转换为2维的网格数据。但如果说社交网络数据有60亿个节点,那其完整的邻接矩阵就是60亿*60亿,不容易表示。而且往往是稀疏矩阵,矩阵里大部分都是0。
- 所以说图其实是一个类型的对象,其中各种关系要复杂的多。如何学习这类对象是一个复杂的任务
更多的graph数据
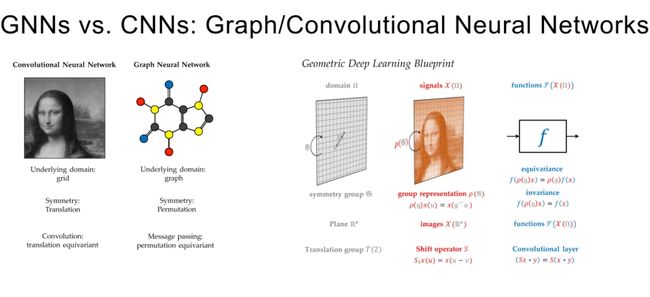
CNN vs GNN:
一个核心点就是CNN处理的图像(二维网格数据),是有一个平移对称的感觉在里面的,里面都是一个个的像素点。而GNN处理的图(graph),存在一个排列对称性,就是说不管你这个图长的什么奇形怪状七扭八扭的,只要都是一样的节点和边,那这个图就是一样的。
某种意义上来说,graph可以看作成grid(二维网格数据)的简单概括,它表示了有关系的像素点(网格点),可以这么理解。
- 卷积神经网络:
- 基础领域:网格。在卷积神经网络中,输入数据(如图像)通常被表示为一个网格,其中每个网格点(即像素)包含一些特定的信息(如颜色)。
- 对称性:平移。卷积神经网络具有平移对称性,这意味着无论图像的对象在何处出现(例如,靠近图像的左边缘、右边缘或中心),网络都能以同样的方式检测到它。
- 卷积:平移等变。这是指,卷积操作在平移下保持不变,即如果输入数据平移,那么输出数据也将以相同的方式平移。
- 图神经网络:
- 基础领域:图。在图神经网络中,输入数据被表示为一个图,其中每个节点包含一些特定的信息,每条边表示两个节点之间的关系。
- 对称性:排列。图神经网络具有排列对称性,这意味着无论图中的节点如何重新排列(即,无论节点的标识如何改变),网络都将产生相同的输出。
- 消息传递:排列等变。这是指,消息传递操作在排列下保持不变,即如果输入数据的节点被重新排列,那么输出数据也将以相同的方式重新排列。
引出GNN
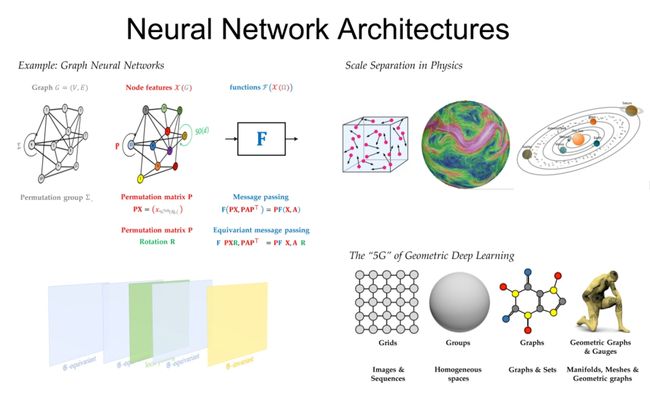
对于graph来说,其每个节点也是有属性的,就像CNN(grid数据)里的一样,每个像素点有相应的像素值。graph里的每个节点也有对应的属性,比如如果图是分子图,那么里面,原子(node)可能是亲水性的或者疏水性的。如果图是社交网络,那么人(node)就有不同的爱好/兴趣。这些都可以作为一个向量进行表示其节点属性。我们要做的任务之一就是研究这些节点属性。
GNN要做到的一个任务(挑战)就是,因为是图,对于同一个图的不同形式的输入要有相同的结果输出
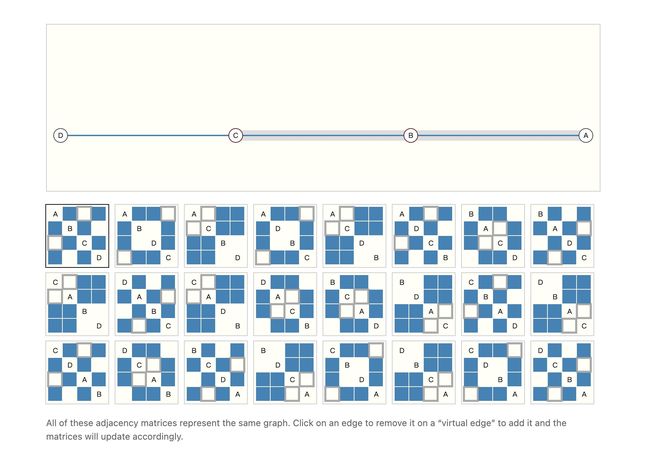
比如如下24种矩阵表示的是同一个图,但是输入的矩阵不一样(表示形式),我们通过GNN得到的结果应该是一样的。(因为本质是同一个图)

主流的一些神经网络结构,我们今天主要是学GNN
2. Graph Neural Networks
Machine Learning Lifecycle
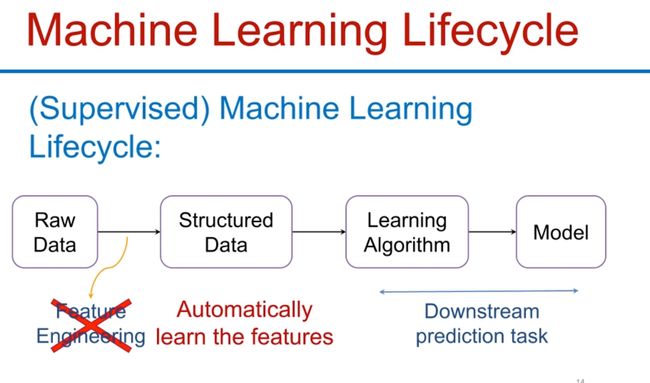
机器学习生命周期如图所示,传统的算法要自己做特征工程,将数据预处理为结构化数据,现代的算法(深度学习)则可以自己学习特征,直接喂没加工的数据就行,这也就是所谓的表征学习(Representation Learning)
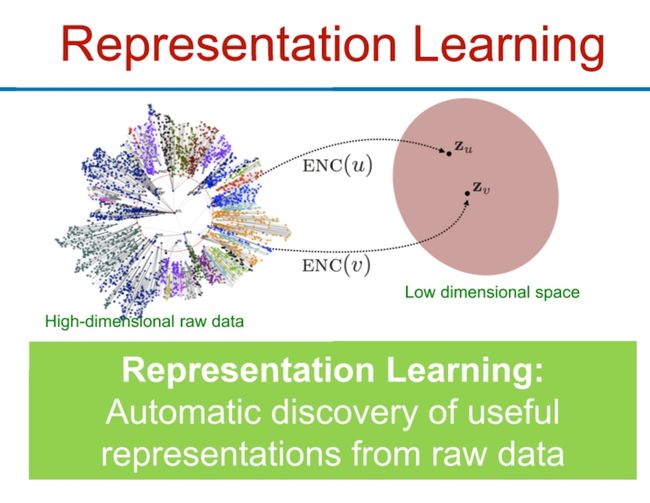
在encoder(编码器)中,将不同的元素embed(映射)到嵌入空间里。这就是Representation存在的地方。比如以前nlp里讲过的一种词嵌入(word embedding)技术(word2vec),就是这种东西。
learning graph is hard
现代的深度学习的算法,都是设计服务于简单的序列/网格数据,但是像图(graph)这样的数据是无法简单的用序列/网格表示的,所以我们需要更广泛应用的深度学习神经网络。
但是这个过程并不容易,因为graph太复杂了,复杂性和动态性,为什么呢:
- 任意大小和复杂的拓扑结构:网络的规模和形状是任意的,而且它们的拓扑结构往往比网格更复杂。例如,在图像(即网格)中,每个像素都有固定的邻居,但在网络中,节点的邻居数量和身份可能会因节点而异。此外,网络中没有像网格中那样的空间局部性,这使得不能直接应用许多针对网格的技术(如卷积)。
- 无固定的节点顺序或参考点:网络中的节点没有固定的顺序,也没有固定的参考点。这意味着不能仅仅根据节点的标识或位置来推断出它的特性或行为。
- 常常是动态的并有多模态特性:许多网络都是动态的,即它们的结构和属性随时间变化。例如,在社交网络中,新的连接可能会形成,老的连接可能会消失,而节点的属性(如用户的兴趣)也可能会改变。此外,网络的节点和边可能会有多种类型的属性,这些属性可能会以多种模态存在(如数值、文本、图像等)。
Feature Learning in Graphs
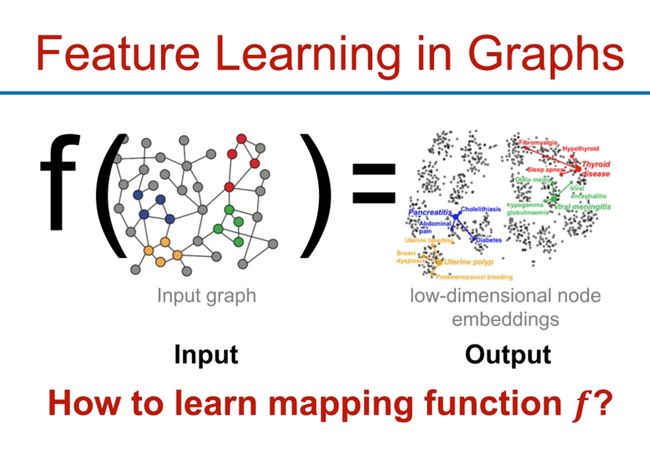
还是需要依靠embed技术(嵌入),就像我们在前面的LLM里学到的Attention机制一样,是能捕捉词语的上下文关系一样,节点与其邻居也有这样的关系,我们在节点本身的属性基础上,还需要获得其与周边节点的关系信息。这是graph的特征学习。输入是图,输出是一个低维空间,比如二维平面,每个节点表示是一个坐标(举例子,不一定准确)。
在图中的特征学习主要是指如何从图结构中提取有用的信息并将其转换为能够用于后续机器学习任务的特征表示或嵌入。这通常涉及学习一个映射函数f,该函数能够将图中的节点(或边)映射到一个低维空间,这个低维空间(也称为嵌入空间)通常具有比原始图结构更容易处理的性质。
在这个过程中,输入是图结构,输出是低维节点嵌入。例如,在医疗领域,输入图可能代表疾病的网络,其中节点代表不同的疾病,边代表疾病之间的关系(如共发性)。通过图的特征学习,我们可以得到每种疾病的低维嵌入,这些嵌入捕捉了疾病之间的关系并可以用于后续的机器学习任务,如疾病预测或药物推荐。
如何学习映射函数f是一个关键的问题。常见的方法包括图卷积网络(Graph Convolutional Networks)和图注意力网络(Graph Attention Networks)等。这些方法都试图通过利用图的拓扑结构和节点特征,学习出能够捕捉节点之间关系的嵌入。
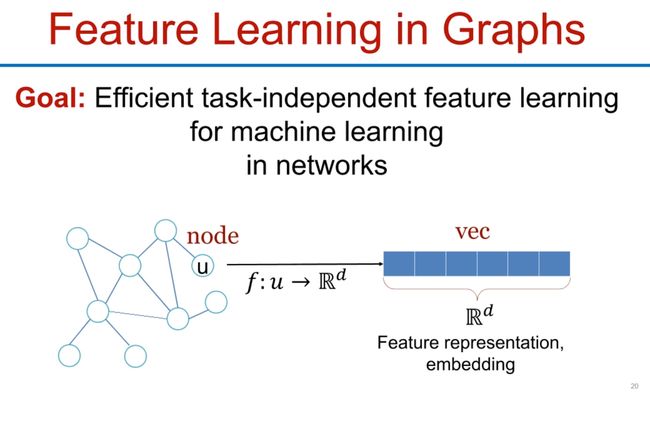
将一个节点转换成一个对应的向量,图嵌入技术。
目标是为网络中的机器学习任务提供有效的、独立于任务的特征学习。换句话说,我们希望学习到的特征或嵌入不仅对当前的任务有用,而且能够轻松地迁移到其他相关的任务上。例如,学习到的疾病嵌入不仅可以用于预测某种疾病的发病率,还可以用于推荐针对这种疾病的治疗方法。
这里补充一个小知识点,就是关于embed(嵌入)这个词,在深度学习里老是见到:
在机器学习中,"embed"或者"embedding"一般有两个主要的含义:
- **表示转换或映射:**这个意思源于词嵌入(word embedding)的概念,例如Word2Vec、GloVe等。这里的"embed"意味着将词语或其他类型的对象从原始的表示空间映射到一个新的(通常是低维度的)表示空间。比如,我们可能将一个单词(通常以one-hot编码的方式表示)映射到一个实数向量空间(例如,一个300维的向量)。这种新的表示空间被设计为能捕获到单词之间的某些关系(如语义或语法相似性)。
- **数据的嵌入式表示:**在更广泛的意义上,"embed"或"embedding"也可以指的是任何将数据从一个表示空间转换到另一个表示空间的过程。这可以涵盖词嵌入之外的其他类型的嵌入,例如实体嵌入(entity embedding)、图嵌入(graph embedding)等。
Ways to Analyze Networks

这是分析网络的一些方式和任务:
- 节点分类(Node Classification):在这种任务中,目标是预测给定节点的类型。例如,在社交网络中,我们可能想要预测一个用户的兴趣或职业。
- 链接预测(Link Prediction):这种任务的目标是预测两个节点之间是否存在链接。例如,在一个社交网络中,我们可能想要预测两个用户是否将成为朋友。
- 社区检测(Community Detection):社区检测的目标是识别出节点之间的紧密链接集群,这些集群可以被视为网络中的社区或群组。例如,在社交网络中,我们可能想要找出具有相似兴趣或背景的用户群体。
- 网络相似度(Network Similarity):在这种分析中,目标是衡量两个节点或两个网络的相似度。这可能包括比较节点的属性、结构位置,或者比较整个网络的拓扑结构。
A Naive Approach 介绍一种最初级的学习图的方式
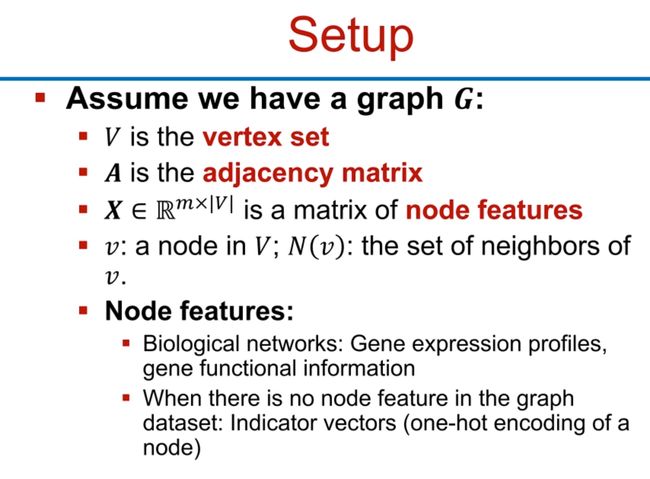
图的符号表示,假设我们有一个图G:
- V是顶点集
- A是邻接矩阵
- X∈R^m*|V| 是节点特征的矩阵,m应该是每个节点的特征数量,比如社交网络里一个人有四个爱好:唱、跳、rap、篮球,那么m=4
- v:V中的一个节点;N(v):节点v的邻居集合
节点特征:
- 在生物网络中,节点特征可能包括基因表达谱和基因功能信息。
- 当图数据集中没有节点特征时,我们可以使用指示器向量(节点的one-hot编码)作为节点特征。

最初级的方法就是利用传统的机器学习思想,将数据转换成结构化数据,投喂到神经网络中。在这里就是将邻接矩阵和特征矩阵结合在一起,然后将它们输入到一个深度神经网络中进行处理。
然而,这个想法存在一些问题:
- 参数过多:深度神经网络需要大量的参数来学习,对于大规模的图,这可能导致计算需求变得非常大,而且可能导致过拟合。
- 不适用于不同大小的图:如果我们有不同大小(即节点数量不同)的图,这种方法可能就不再适用,因为深度神经网络的输入和输出大小是固定的。
- 对节点顺序敏感:由于我们是将邻接矩阵和特征矩阵直接作为输入,这就意味着我们的模型会对节点的顺序敏感。然而,在许多图中,节点的顺序并不重要,因此我们不希望我们的模型对这个顺序敏感。就是前面讲的,我们要对同一个图的不同形式输入(不同节点顺序表示),得到永远相同的输出!!
因此,虽然这种直接的方法在某些情况下可能有效,但在许多实际应用中,我们需要更复杂、更灵活的方法来处理图数据,例如图神经网络(Graph Neural Networks)。
一种更好的处理方式:深度图编码器(Deep Graph Encoders)
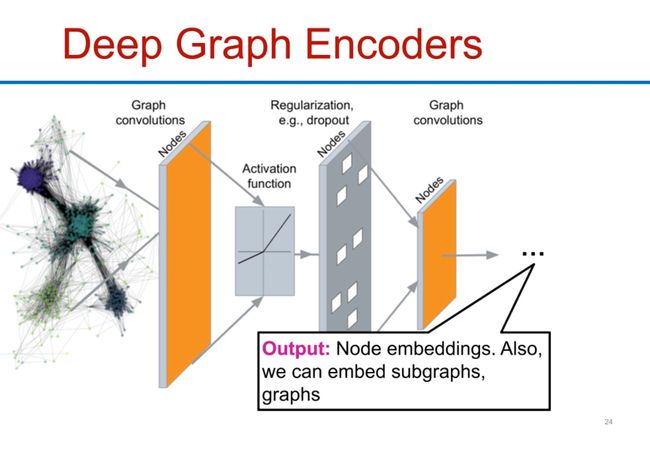
还是推荐大家去这个博客开头玩一下,可交互:A Gentle Introduction to Graph Neural Networks
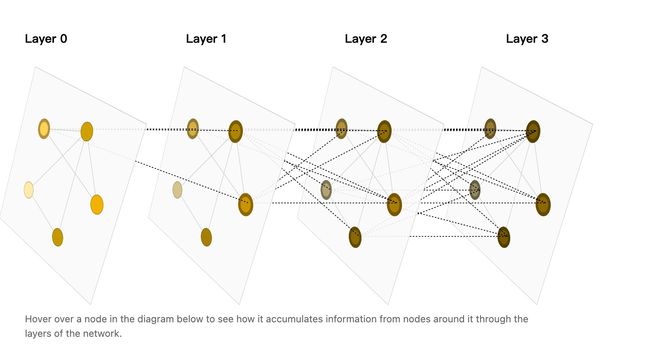
这里更好的方式就是一种类似于卷积的方式,只不过用在图上,对于一个节点来说,它第一层卷积学到的就是它邻居的数据,第二层就是邻居的邻居,第三层就是邻居的邻居的邻居这样。通过这种方式实现图的嵌入,提取出节点的信息。而不是前面简单粗暴的转换成邻接矩阵。
By gpt4:
深度图编码器(Deep Graph Encoders)使用一种特殊的卷积运算——图卷积(Graph Convolutions),来提取节点或图的特征,生成高质量的节点或图嵌入(embeddings)。
下面是一种常见的深度图编码器的架构:
- 图卷积(Graph Convolutions):这是一个操作,用于在图中的每个节点上进行信息聚合。每个节点的新特征是其邻居特征的函数,这个函数通常是加权和或平均。
- 激活函数(Activation Function):通过一个非线性函数,如ReLU或tanh,来增加模型的表达能力。
- 规范化(Regularization):例如,dropout是一种用于防止过拟合的技术,通过在训练过程中随机丢弃一部分节点,来增加模型的泛化能力。
- 图卷积(Graph Convolutions):进行另一轮的图卷积,进一步提取和整合节点特征。
输出结果是节点嵌入,这些嵌入能够捕获节点的特征及其在图中的位置。此外,我们还可以将这种方法扩展,用来生成子图或整个图的嵌入。
这种深度图编码器在处理具有复杂结构和特征的图数据时,比传统的深度学习方法更有效。
- 类比一下CNN的卷积,从image到graph。当然nlp里面也是这种思想,简单来说就是将自身和周围的属性整合到一起啦
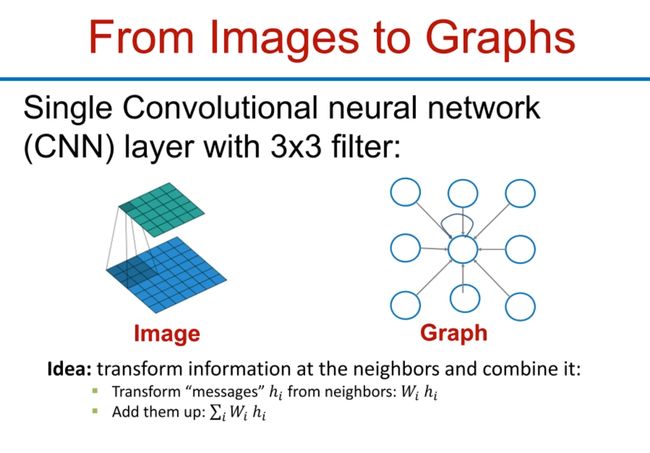
Graph Convolutional Networks(GCN)
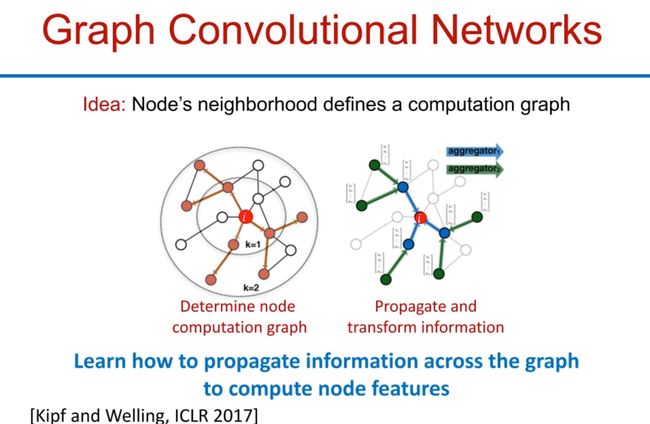
图卷积网络(Graph Convolutional Networks,GCNs)是一种深度学习模型,专门用于处理图数据。GCNs的主要思想是一个节点的邻域定义了一个计算图,我们可以通过这个计算图,传播和转换节点间的信息,以计算节点的特征。
GCNs的操作步骤可以简述如下:
- 确定节点计算图:每个节点及其邻居构成一个计算图。例如,对于一个目标节点,其计算图将包括该节点及其邻居。
- 传播和转换信息:在计算图中,信息将从一个节点传播到另一个节点。同时,每个节点的信息将通过某种转换(例如,通过一个神经网络)进行更新
Idea: Aggregate Neighbors
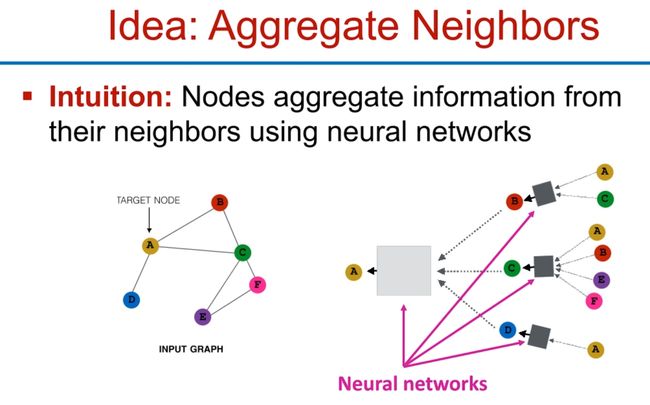
GCNs的一个关键思想是聚合邻居信息,邻域聚合(Neighborhood Aggregation)是图神经网络中的关键概念。不同的方法在如何聚合邻居信息方。基本的直觉是,节点可以通过神经网络从其邻居那里聚合信息。例如,一个节点可能会计算其所有邻居特征的加权和,然后将这个加权和作为输入,通过一个神经网络进行处理,生成新的节点特征。
这里图中可以看到第0层是A,第1层是BCD,第2层也存在A,这个不是循环,是按一层层来的,先有A的信息给了B、C、D更新,然后这个信息再传递到第0层的A。
这种方法的一个优点是,它可以直接处理图数据,而无需将图转换为其他格式。这使得GCNs在处理复杂图数据时,能够保留图的结构和特征信息。
以上所述的GCNs的工作原理是根据Kipf和Welling在2017年的ICLR会议上发布的论文《Semi-Supervised Classification with Graph Convolutional Networks》中的方法
- 大概的过程:
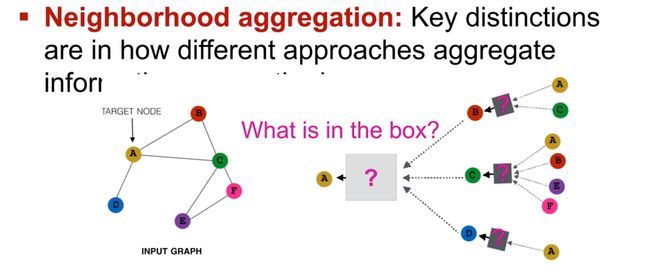
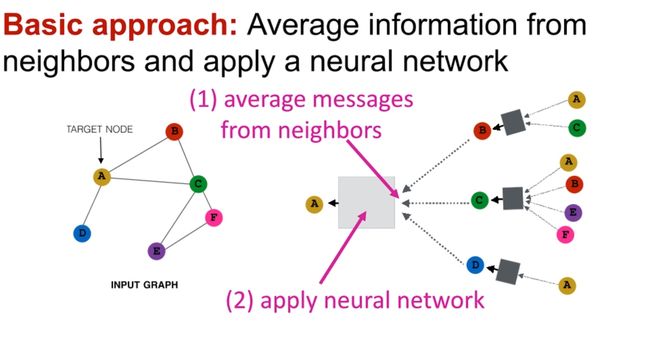
不同的方法在如何聚合邻居信息方面存在一些区别,但基本的方法是从邻居中平均信息,并应用一个神经网络进行处理。具体来说,这个过程可以分为两步:
- 从邻居平均信息:首先,目标节点会从其邻居那里收集信息。这些信息可以是节点特征、边特征,或者是来自更远节点的信息。这些信息被收集起来,并计算其平均值或其他形式的汇总统计。
- 应用神经网络:接下来,将这些汇总统计作为输入,传递给一个神经网络。这个神经网络可以是任何类型的网络,比如一个全连接网络、一个卷积网络,或者是一个更复杂的网络。这个网络的任务是将这些汇总统计转换为新的节点特征
这个从邻居这里收集信息应该不难理解,应用神经网络乍一看有点抽象,但实际上不抽象,一定程度上就好像CNN的卷积+MLP一样。从GCN的角度理解,其实就是根据原来的数据去学习新的节点特征的过程。(语言表达能力有限,反正就是参考CNN)
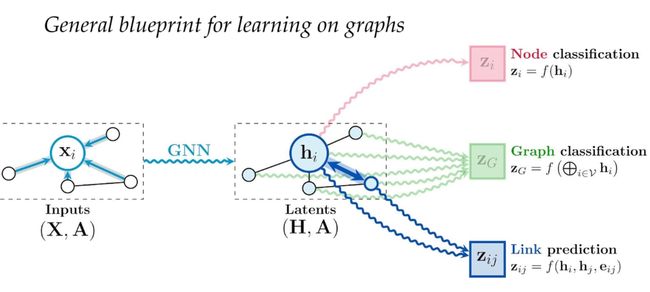
以上的流程形成了学习图数据的一般蓝图,可以用于节点分类、图分类和链接预测等任务:
- 节点分类:对每个节点应用一个函数f,输入是通过图神经网络学习得到的节点特征,输出是节点的类别。
- 图分类:对整个图应用一个函数f,输入是图中所有节点的特征(可能是经过某种汇总的),输出是图的类别。
- 链接预测(边预测):对每对节点应用一个函数f,输入是这两个节点的特征(以及可能的边特征),输出是这两个节点之间是否存在链接的预测。
The three “flavours" of GNN layers

图神经网络(GNN)层的设计是当前非常活跃的研究领域。一个GNN层的主要目标是建立在图形上的排列等变函数F(X, A),通过共享的局部排列不变函数φ(X#) Xw)来实现。在这里,X代表节点特征,A代表邻接矩阵。
以下是一些常用的术语:
- F被称为"GNN层"。
- φ被称为"扩散"、“传播"或"消息传递”。
那么,如何实现φ呢?这是一个非常活跃的研究领域,幸运的是,几乎所有的φ都可以按照三种"风味"(即注意力、消息传递和卷积)进行分类。
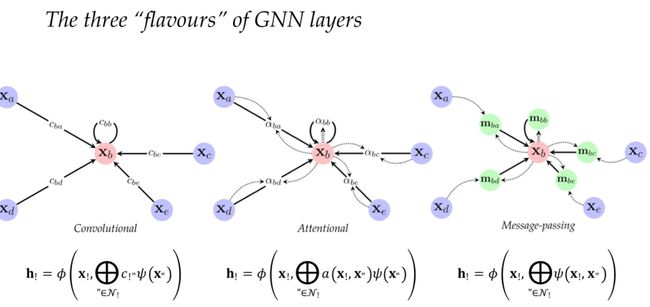
图神经网络(GNN)有三种“风味”的层:
- 卷积(Convolutional):这种类型的图神经网络层受到卷积神经网络的启发,对每个节点和其邻居的特征应用一种变换(通常是线性变换),然后将结果聚合(通常是通过求和或平均)来更新每个节点的状态。这种机制依赖于图的拓扑结构,以一种不依赖于节点的特定排序的方式提取局部模式。Graph Convolutional Networks (GCNs)就是这种类型的一个例子。
- 注意力(Attentional):在这种类型的图神经网络层中,每个节点的更新依赖于其邻居的信息,但每个邻居的贡献是通过注意力机制进行加权的。这意味着一些邻居可能对当前节点的更新有更大的影响,而其他邻居的影响可能较小。(通过注意力机制判断不同邻居的不同影响)。这种机制允许模型学习到在给定上下文中哪些邻居是重要的。Graph Attention Networks (GATs)就是这种类型的一个例子。
- 消息传递(Message-passing):在这种类型的图神经网络层中,每个节点都向其邻居发送“消息”,然后根据收到的所有消息更新自己的状态。这些“消息”是由发送节点的特征生成的,可以通过各种方式(例如,加权平均,最大值池化等)聚合收到的所有消息。这种方式可以帮助模型捕捉到图中的复杂模式和依赖关系。消息传递神经网络**(MPNN)**就是这种类型的一个例子。
这三种类型的图神经网络层都是尝试从节点的邻居中获取信息,但它们采用了不同的策略,因此可能在不同的任务和图结构中表现不同。
我有点分不清GCN和MPNN,就问了一下gpt4,还是不太懂,但是未来有时间慢慢再了解:
图卷积网络(GCN)和消息传递神经网络(MPNN)在某种程度上有相似之处,因为它们都试图在节点与其邻居之间传递信息。然而,它们在实现细节和概念上是有所不同的。
- 图卷积网络(GCN):GCN受到卷积神经网络(CNN)的启发,它试图从一个节点的邻居捕获局部信息。与CNN在规则网格上进行局部卷积不同,GCN在不规则的图结构上进行卷积。在GCN中,每个节点的新特征是其邻居特征的加权平均(加权项通常是与边相关的属性或节点的度),然后再通过一个非线性激活函数。这种操作可以视为一种形式的卷积,它在图的空间结构上定义了一种局部邻域。
- 消息传递神经网络(MPNN):MPNN的工作方式是每个节点向其邻居发送“消息”,然后根据收到的所有消息更新自己的状态。这些消息是通过神经网络函数从发送节点的特征生成的。然后,节点将收到的所有消息聚合(比如通过求和或取最大值),并更新自己的状态。这种方式使得模型可以捕捉到更复杂的依赖关系,因为每个节点都可以根据它自己的特征以及收到的消息来决定如何更新自己的状态。
简单来说,卷积操作侧重于捕获节点和其邻居之间的一种固定模式(即“平均”),而消息传递则允许更复杂的交互模式,因为它允许每个节点根据它自己的特征和收到的消息来自主地更新其状态。
3. Deep Learning on Graphs
Deep Encoder
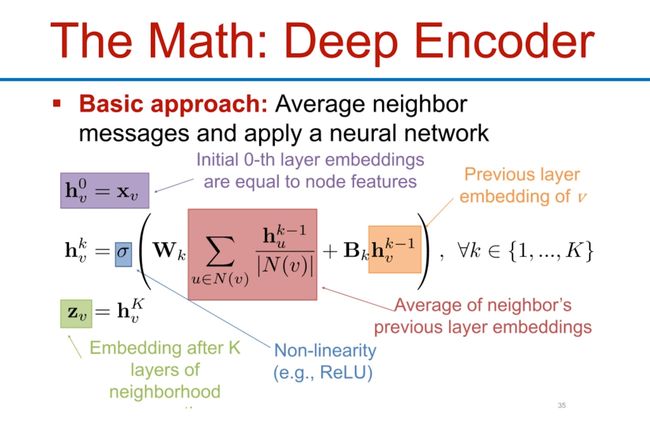
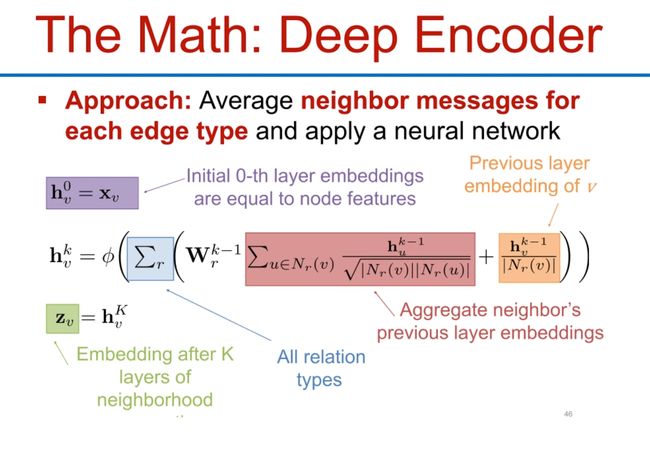
这个数学模型描述的是深度编码器(Deep Encoder)在图神经网络中如何工作。这种深度编码器的基本方法是采用邻居节点的信息(即消息),对其进行平均,并应用一个神经网络。
以下是对这个模型的具体解释:
- 初始的嵌入:初始时刻,即0层(0-th layer),每个节点的嵌入(也称为特征向量)就是其本身的特征( X i X_i Xi)。
- 计算邻居的平均嵌入:对于每一个节点 v v v,它的邻居节点集合记为 N ( v ) N(v) N(v)。对于这个节点集合中的每一个节点u,取出其上一层的嵌入(也就是 h u k − 1 h^{k-1}_u huk−1),并计算这些嵌入的平均值。
- 应用神经网络:然后,将这个平均嵌入乘以一个权重矩阵 W k W^k Wk,加上一个偏置项 B k B^k Bk,并通过一个非线性激活函数 σ \sigma σ(如ReLU),得到节点u在第k层的嵌入 h u k h^k_u huk。
以上步骤在k层中进行,每一层都在原来的嵌入上增加了更多的邻居信息,最终生成深层次的节点表示。
就类似前面的这个过程,总共有k层:
- 模型参数
模型参数就是权重和偏置:

参数设置:
- 可训练的权重矩阵:在每一层的计算过程中,都有一个权重矩阵W_k用于更新节点的嵌入。这个权重矩阵是模型需要学习的参数之一。每一层的权重矩阵可以不同,也就是说W_k可以随着层级k的变化而变化。
- 计算节点嵌入:每一个节点u的嵌入h(k)_u是通过对其邻居节点在上一层的嵌入求平均,然后乘以权重矩阵W_k,并添加一个偏置项b(k),最后通过一个非线性激活函数计算得到的。
- 损失函数和优化:得到的节点嵌入可以用于计算任何适当的损失函数,例如用于节点分类任务的交叉熵损失,或者用于链接预测的负对数似然损失等。然后,通过随机梯度下降(SGD)或其它优化算法来调整权重矩阵和偏置项,使得损失函数最小,从而训练模型。
GraphSAGE


GraphSAGE(图采样与聚合)是一种用于在大型图中进行表示学习的框架。其目的是为了生成节点嵌入,这些嵌入能够在训练时候未见过的节点和图形上进行泛化,这对于许多实际应用非常有价值,因为实际的图经常是动态生成和扩展的。
到目前为止,我们已经通过取邻居消息的(加权)平均来进行聚合,GraphSAGE提出了一种改进的方式。
GraphSAGE方法的关键思想是,对于每个节点,它会先采样一个固定数量的邻居节点,然后将这些邻居节点的特征进行某种形式的聚合(例如,取平均、池化、或者使用LSTM等),然后将聚合得到的邻居特征与该节点自身的特征进行合并(例如,拼接或者求和),并通过一个非线性转换(例如,ReLU)生成新的节点特征。这个过程可以重复多次,类似于在图上进行多层卷积。最后,得到的节点特征就可以用于各种下游任务,例如,节点分类、链接预测等。

三种聚合变体方式:
- 平均:对邻居节点取加权平均 公式中的AGG表示聚合函数,其中hk-1是前一层的嵌入,u属于N(y),其中N(y)表示节点y的邻居集合。
- Pool(池化):对邻居向量进行转换并应用对称向量函数 比如,可以对每个邻居向量进行线性变换(乘以权重矩阵Q),然后在所有邻居向量上取元素级的平均值或最大值。这种方法的一个好处是,因为平均值和最大值操作都是对称的,所以无论邻居节点的顺序如何,聚合结果都是一样的。
- LSTM:将LSTM应用到邻居的重新排序上 对于每个节点,首先对其邻居节点的嵌入进行重新排序,然后将这些嵌入作为序列输入到一个LSTM网络中。得到的LSTM的最后输出被用作该节点的新的嵌入。这种方法的一个缺点是,LSTM不是对称的,所以同样的邻居节点,如果顺序不同,可能会产生不同的聚合结果。
Application to Drug Design/Evaluation
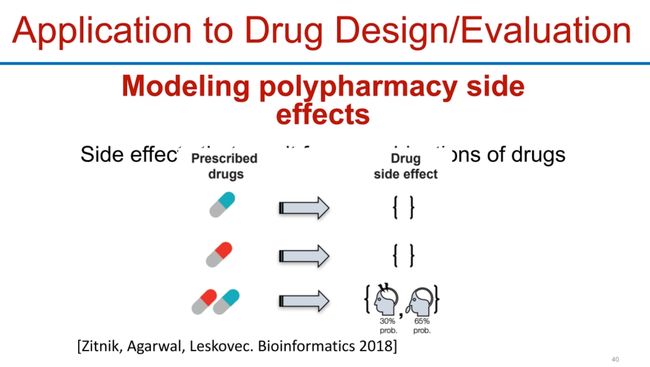
模型应用在多药治疗的副作用预测中: 给定一组被处方的药物,该模型能预测这些药物可能会引起的副作用。每种副作用以一个概率表示,概率越高,产生该副作用的可能性越大。
在药物的研究和开发过程中,理解和预测药物的副作用是至关重要的。如果一个药物的副作用过于严重,那么这个药物可能就无法投入市场。通过使用图神经网络来预测药物的副作用,研究人员可以更好地理解药物之间的相互作用,并可能帮助他们设计出更安全、更有效的新药。
Heterogenous Networks
- 异构网络,就是一些网络的节点和边具有不同的属性,这样子问题就更加复杂一些
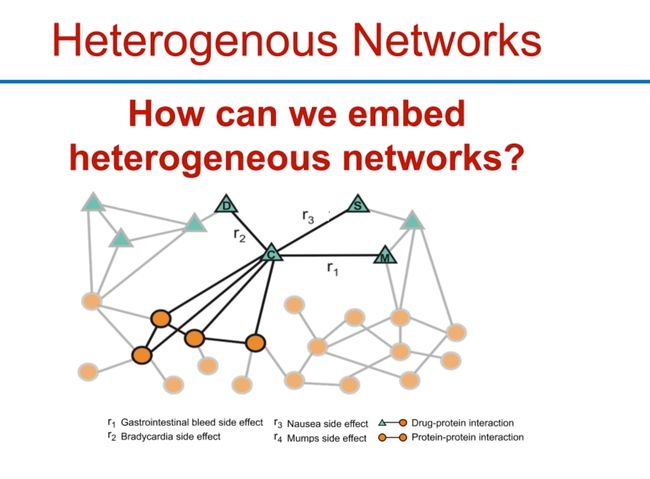
By gpt4:
异构网络如何嵌入?
异构网络是指网络中的节点和边可以有多种类型。例如,在社交网络中,节点可能代表个人或组织,边可能代表友谊或商业关系。在这种情况下,我们需要一种方法来嵌入这种多样性的网络。
对异构网络进行嵌入的一种方法是使用图神经网络(GNN)。这些模型可以处理多种类型的节点和边,**通过考虑节点和边的类型信息来生成节点的嵌入。**例如,具有不同类型边的节点可能会被编码为具有不同的特征。
对于每种类型的节点或边,我们可以有一个不同的嵌入模型或一组参数。然后,通过合适的聚合策略(例如,加权求和,平均,最大池化等)来组合不同类型的邻居信息。这就是所谓的异构图神经网络。
异构图神经网络的设计和应用是当前图深度学习研究的热点,具有丰富的理论研究和应用实践。
- setup

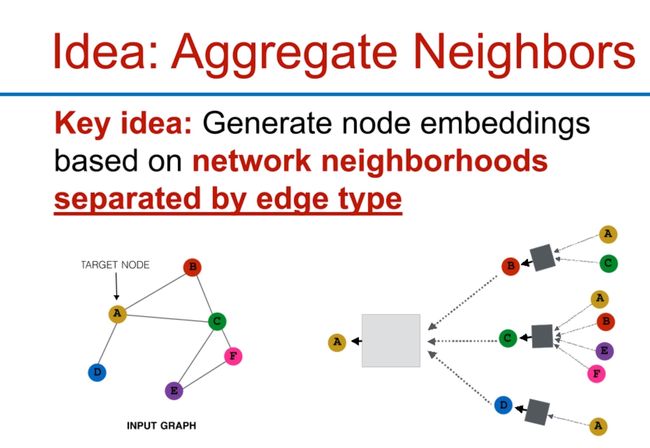
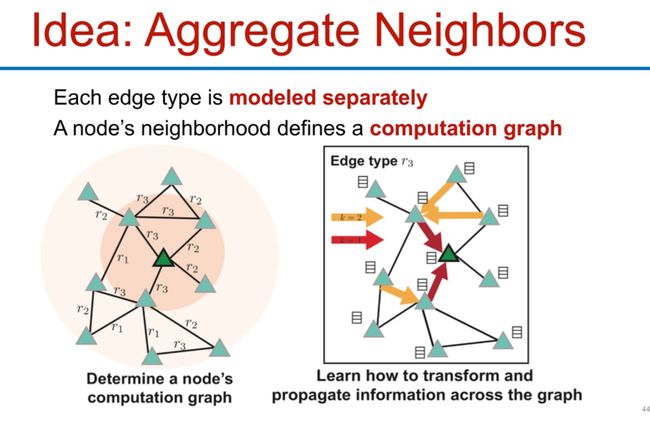
- 感觉边的类型进行邻居的聚合:
- 关键思想:根据网络邻域生成节点嵌入分隔边的类型
- 每种边类型都单独建模节点的邻域定义了一个计算图
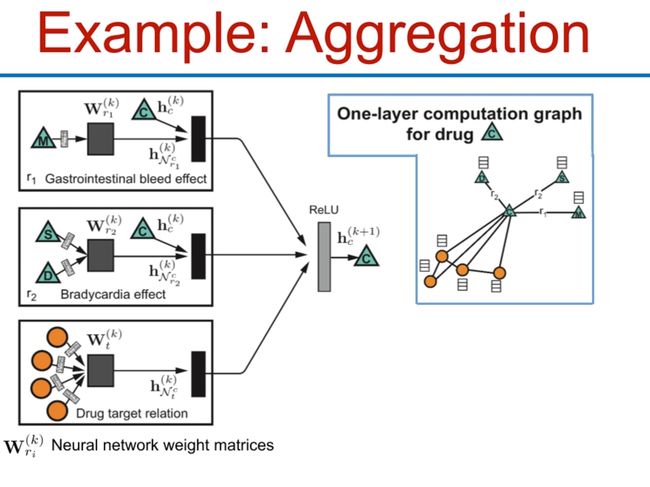
- 数学公式
Training the Model
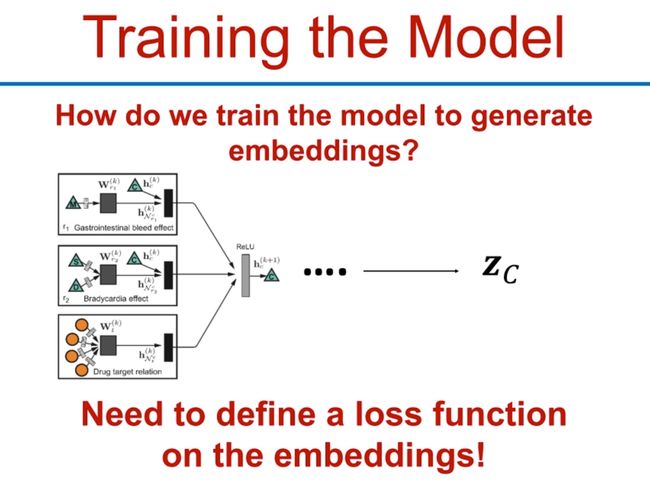
我们需要去定义一个损失函数,对embedding后的内容进行训练
4. GNNs for Protein folding
Chemical Structures as Graphs

- 化学结构作为图: 化学分子的结构可以看作是图,其中原子被看作是顶点,化学键被看作是边。这种视角为化学分子的计算和分析提供了强大的工具,特别是在计算化学和药物设计等领域。
- 分子的结构相似性: 分子的结构相似性是通过比较两个或更多分子的化学结构来度量的。这种相似性通常是通过比较分子图的特性,如原子类型,化学键类型,原子间的连接关系等来确定的。
- 图论与化学: 图论是数学的一个分支,主要研究图的属性和结构。在化学中,图论被广泛应用于分子结构的描述和分析。通过将分子视为图,可以使用图论的概念和技术来解决许多化学问题,比如判断两个分子是否同构,预测分子的化学反应性等。
- Weisfeiler-Lehman测试: Weisfeiler-Lehman (WL) 测试是一种用于确定两个图是否同构的算法。同构的图在结构上是相同的,即使顶点的标签或排列顺序不同。WL测试通过一种迭代的过程来对图的顶点进行重新标记,如果两个图在经过足够多次迭代后得到的标记相同,那么这两个图就被认为是同构的。在化学中,WL测试可以被用来比较分子的结构,从而帮助化学家识别结构相似的分子。
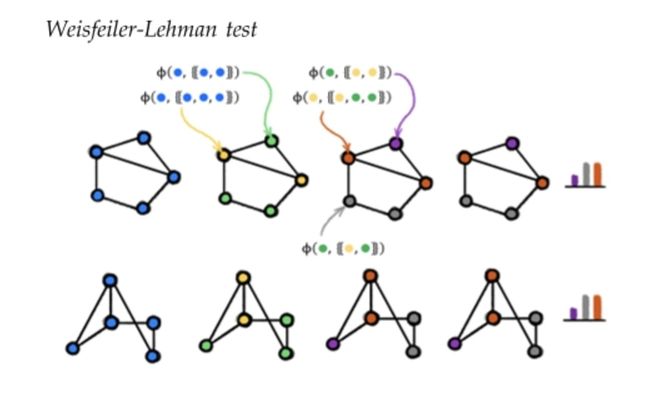
简单解释一下,我们看第一排:
- 第一张图蓝色的,所有节点一样
- 我们把有两个邻居的节点标为绿色,有三个邻居的节点标为黄色,我们就得到了第二张图
- 然后,我们把有两个黄色邻居的节点标为紫色,把有二绿一黄邻居的节点标为橙色,把有一绿一黄邻居的节点标为灰色
- 最后我们统计这个分子图的节点就是一紫二灰二橙。
- 我们发现下面这个分子图虽然长的跟上面这个不一样,但是通过这样简单计算出来也是一紫二灰二橙。我们就认为是同构的。
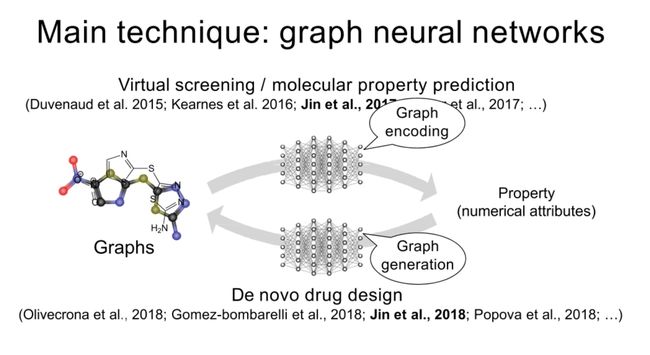
这段内容概述了利用图神经网络进行药物研发的两个主要技术:虚拟筛选/分子属性预测和新药设计。
虚拟筛选/分子属性预测: 虚拟筛选是一种计算技术,用于在大规模化合物库中识别可能的药物候选物。在这个过程中,图神经网络被用于学习分子的低维度表示,这些表示可以用于预测分子的属性,如溶解度、毒性等。这项技术已经在多项研究中得到应用,包括Duvenaud等人(2015),Kearnes等人(2016)和Jin等人(2018)的研究。
新药设计: 新药设计,也被称为"de novo"药物设计,是一个利用计算工具来设计新的潜在药物的过程。在这个过程中,图神经网络被用于生成新的分子结构。这项技术已经在多项研究中得到应用,包括Olivecrona等人(2018),Gomez-bombarelli等人(2018),Jin等人(2018)和Popova等人(2018)的研究
Protein Structure Prediction
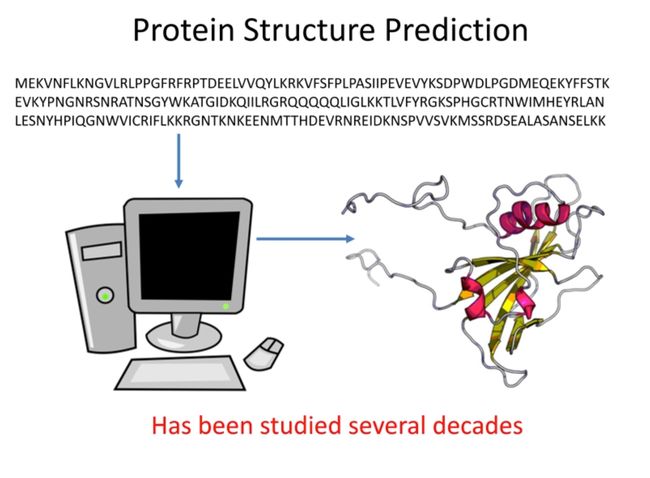
三维的蛋白质结构由一维的氨基酸序列折叠而成,这个问题已经被研究了几十年。
蛋白质结构预测是一种用于确定蛋白质的三维结构的方法,通常是通过其氨基酸序列。理解蛋白质的三维结构是非常重要的,因为它可以帮助我们理解蛋白质的功能,并在药物设计中找到可能的药物靶点。
蛋白质折叠通常涉及如下几个主要的结构元素:
- α-螺旋(Alpha-helices): α-螺旋是蛋白质的一种常见二级结构,由于胺基和羧基之间的氢键作用,氨基酸链在空间中旋转形成螺旋状。
- β-折叠(Beta-sheets): β-折叠是蛋白质的另一种常见二级结构,由两个或更多的平行或反平行的β链通过氢键连接在一起形成。
蛋白质结构预测在计算生物学中是一个重要且具有挑战性的问题。许多不同的技术和方法已经被开发出来解决这个问题,包括模板匹配、同源建模,以及更先进的方法,如深度学习。深度学习的方法,例如AlphaFold,已经在CASB (Critical Assessment of protein Structure Prediction)竞赛中取得了显著的成果,显示出其在预测蛋白质结构方面的潜力。
Methods for Protein Structure Prediction
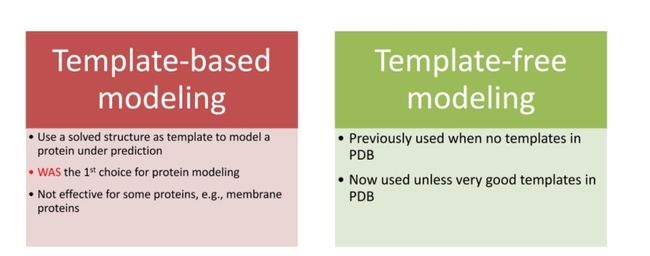
模板依赖建模和模板自由建模是两种常用的蛋白质结构预测方法。
- 模板依赖建模(Template-based modeling, TBM):这种方法依赖于已解决的蛋白质结构数据库(如PDB)中的已知蛋白质结构,将其作为模板。如果一个新的蛋白质序列与已知结构的蛋白质序列高度相似(同源),那么就可以使用这个已知的结构作为模板,预测新的蛋白质的结构。然而,对于一些没有相似模板的蛋白质,例如某些膜蛋白,此方法可能效果不佳。
- 模板自由建模(Template-free modeling, also known as ab-initio or de novo modeling):这种方法不依赖于已知的蛋白质结构模板,而是通过理论模型和计算方法预测蛋白质的结构。例如,这可能包括对蛋白质的物理和化学性质的模拟,如力学模型和电磁模型,或者使用蒙特卡洛方法进行随机搜索。然而,这种方法的计算复杂性通常较高,且预测准确性可能不如模板依赖建模。
最近,深度学习方法(如AlphaFold)已经在蛋白质结构预测中显示出很大的潜力,这种方法既可以利用已知的蛋白质结构信息(如果可用),也可以从头开始预测蛋白质的结构,从而结合了模板依赖和模板自由建模的优点
Old method: fragment assembly
以前使用超级计算机去模拟,需要很大的算力而且成功率很低
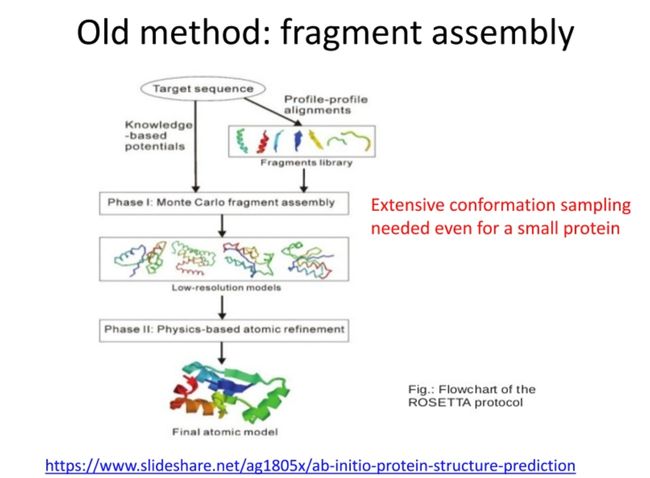
Fragment assembly是一种传统的蛋白质结构预测方法,具体过程如下:
1. 目标序列:首先,我们有一个需要预测结构的蛋白质目标序列。
2. 片段库 (Fragment Library):通过从已知结构的蛋白质中提取短序列片段来创建一个片段库。这些片段的长度通常为3到9个氨基酸。
3. Profile-Profile Alignment:然后,将目标序列的氨基酸剖面与库中的每个片段剖面进行对比。
4. 蒙特卡洛片段装配 (Monte Carlo Fragment Assembly):通过蒙特卡洛方法从片段库中随机选取片段,并将它们拼接在一起以构建蛋白质的初步模型。通过重复这个过程,产生大量的蛋白质模型。
5. 基于知识的势能 (Knowledge-based potentials):使用基于统计的得分函数(如Rosetta的得分函数)来评估和排列这些模型,选取得分最高的模型作为最佳模型。
6. 基于物理的原子精修 (Physics-based atomic refinement):最后,基于物理的能量最小化过程对选定的模型进行精修,包括优化氨基酸侧链位置和小幅度调整蛋白质的主链。
这种方法的一个主要问题是计算效率较低,且准确度受到已知结构片段库的限制。
New Strategy

这个新策略包含了以下步骤:
- 多序列比对(Multiple Sequence Alignment):多序列比对是一种基于序列比对的方法,它用于确定一组蛋白质序列或者核酸序列之间的相似性。通过比对多个序列,我们可以找出共享的进化保守区域,即那些在进化过程中保持不变的区域。这些保守区域通常对于蛋白质的功能和结构至关重要。
- 共演化分析(Co-evolution Analysis):这是一种寻找蛋白质内部氨基酸间联系的方法,即某个氨基酸位置的变化可能会引起另一个氨基酸位置的变化。这种共演化信息可用于预测蛋白质的三维结构,因为在蛋白质中,共演化的氨基酸对往往在空间中靠近。
- 深度神经网络预测相互作用矩阵:输入的数据包括上述的多序列比对和共演化分析结果,以及其他可能的蛋白质序列信息。这个深度神经网络的目标是预测一个相互作用矩阵,其中每个元素表示蛋白质序列中两个氨基酸在空间中的相互作用强度。
- 深度神经网络预测局部结构:类似地,也可以使用深度神经网络预测蛋白质的局部结构信息,比如二级结构(alpha螺旋,beta折叠等)和每个氨基酸的位置。
- 最小化分子力场(Minimization Molecular Force field):在获取预测的相互作用矩阵(二维)和局部结构信息(线性)后,可以使用分子模拟方法(如力场最小化或分子动力学模拟)来生成蛋白质的三维结构。这一步的目标是找到一个蛋白质结构,该结构最好地满足预测的相互作用和局部结构信息。
这个策略提供了一种有效的方法来预测蛋白质结构,与传统的方法相比,它更多地利用了深度学习和序列演化信息,因此通常可以得到更准确的预测结果。
Co-evolution Analysis
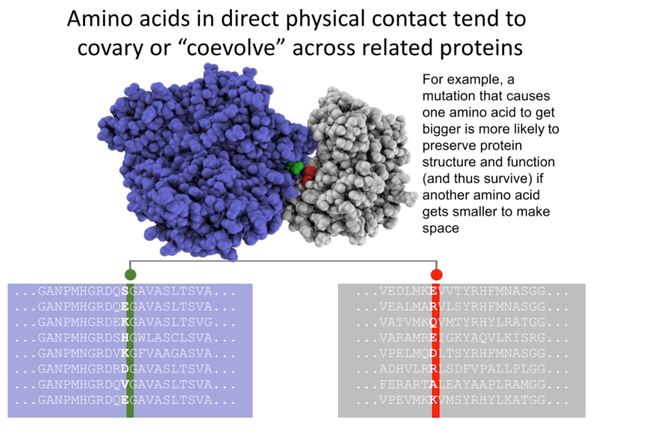
蛋白质的结构和功能严重依赖于其氨基酸的排列顺序和化学性质。如果一个氨基酸突变,使得其边链增大,这可能会干扰蛋白质的结构或者影响其功能,因为这可能会使其与邻近氨基酸的相互作用发生改变。
但是,如果另一个邻近的氨基酸同时发生突变,使其变小,这就可能可以平衡边链的大小变化,保持蛋白质的稳定性。这种情况下,两个氨基酸就会表现出共演化的特性,即它们的变化是协调的。
这种共演化现象可以帮助我们理解蛋白质的三维结构和功能,因为共演化的氨基酸对通常在三维结构中紧密接触,共同参与形成蛋白质的活性位点或者结构域。因此,通过分析多个蛋白质序列的共演化模式,我们可以预测蛋白质的结构或者功能位点。
共演化的概念可能比较抽象,我们通过一个简单的例子来理解它。
假设我们有一种具有三个氨基酸的极简单的生物体。这三个氨基酸排列在一条链上,形成了一种蛋白质。我们将这三个氨基酸分别命名为A,B,C。
在一种理想的环境下,这三个氨基酸的理想形态分别为大,中,小。也就是说,最好的蛋白质形式是大的A,中的B,小的C。这种组合能让生物体达到最好的适应环境的效果。
然而,生物体在演化过程中会发生突变,氨基酸可能会改变形态。比如,A可能突变为中型,或者B突变为大型。
现在,如果A突变为中型,那么与A相邻的B就会面临压力,因为它现在与A一样大了。为了维持蛋白质的最优形态,B可能也会随之突变为小型。这样,A和B就通过共同适应环境压力而发生了共演化。
如果我们只看一个生物体,可能很难观察到这种共演化的现象。但如果我们观察很多具有相似蛋白质的生物体,我们就可以看到某些氨基酸位置上的突变是有关联的。例如,我们可能会观察到,每当A变为中型时,B也往往变为小型。
这就是共演化的基本概念。在实际的生物体和蛋白质中,情况会复杂得多,因为一个蛋白质可能由数百或数千个氨基酸组成,每个氨基酸可能以20种不同的形式出现。但即使在这种复杂的环境中,我们仍然可以通过统计分析发现共演化的模式,从而预测蛋白质的结构或功能。
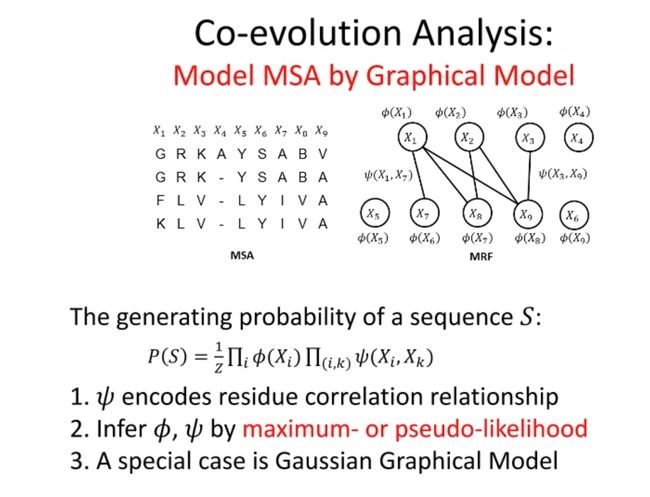
这里是对多序列比对(MSA,Multiple Sequence Alignment)的共演化分析的介绍。共演化分析在生物信息学中被广泛应用,尤其是在蛋白质结构预测和功能位点预测等领域。
这里的图形模型(也被称为图模型或马尔可夫随机场,MRF)是用来建模MSA的。它将氨基酸序列中的每个位置看作一个随机变量,并假定两个位置之间的关系可以由一个权重函数来描述。
对于给定的氨基酸序列S,其生成概率 P ( S ) P(S) P(S)可以由这个图模型来计算,其中 φ ( x ) φ(x) φ(x)表示序列中某个位置的单点概率,而 w ( x i , x j ) w(x_i, x_j) w(xi,xj)表示序列中两个位置的相关性或协变性。
这个模型的参数 φ φ φ和 w w w可以通过最大似然法或伪似然法进行估计。这种方法的一个特例是高斯图形模型,它假定随机变量服从多元正态分布。
总的来说,这个方法的目标是通过分析蛋白质序列的共演化模式,来预测蛋白质的结构或功能位点。

在蛋白质结构预测中,通常需要收集并利用一些蛋白质的特征和标签。这些特征和标签可以帮助我们理解蛋白质的性质并预测其未来的行为。
- 序列特征(Sequential Features): 这是指蛋白质序列中的信息,如保守性分布(conservation profile)和预测的局部结构(predicted local structure)。保守性是指某个位点的氨基酸在不同物种或不同蛋白质家族中的变异程度。预测的局部结构是指蛋白质中每个氨基酸可能形成的二级结构类型(如α-螺旋,β-折叠)。
- 配对特征(Pairwise Features): 这是指基于两个氨基酸之间关系的特征,包括相互信息(mutual information),直接共演化(direct co-evolution),接触势(contact potential)等。相互信息和共演化是量化两个位点氨基酸变化依赖性的方法,接触势则是根据氨基酸类型预测它们之间可能发生物理接触的概率。
- 标签(Labels): 这是我们想要预测的目标,例如氨基酸间的距离。这个距离通常被分为几个区间,比如小于8埃,8-15埃,大于15埃。值得注意的是,这种标签分布通常是不平衡的,因为相距较远的氨基酸对(大于15埃)和相距较近的氨基酸对(小于8埃)在蛋白质中的数量通常要多于中间距离的氨基酸对。
在收集了这些特征和标签后,我们就可以使用一些机器学习或深度学习的方法来训练一个模型,该模型可以从这些特征中学习并预测蛋白质的结构或者行为。
Towards An End-to-End Workflow
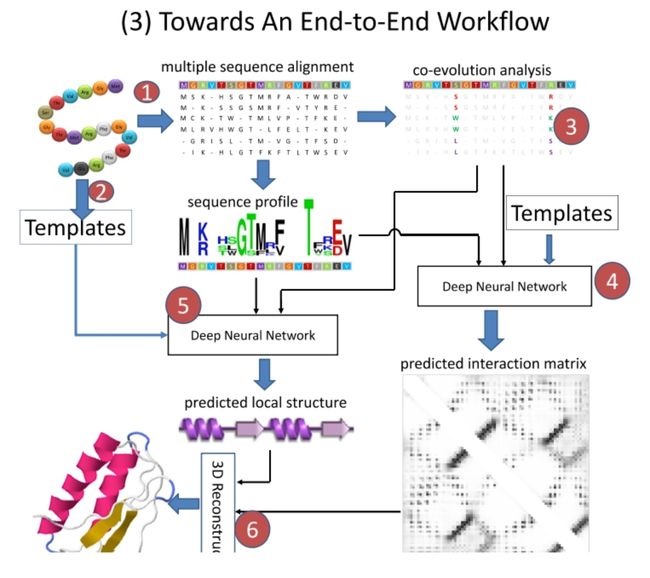
"Towards An End-to-End Workflow"工作流程引入了一个额外的步骤,即"Templates"步骤。
Templates步骤通常涉及到使用已知的蛋白质结构作为模板,这些模板在某种程度上与目标蛋白质的序列相似。这些模板可以提供额外的结构信息,有助于提高预测的准确性。这种方法尤其对于那些能找到良好模板的蛋白质序列特别有用。
在这个工作流程中,模板不仅被用于初始化预测,还被用于训练深度神经网络。这意味着,网络不仅从多序列比对和共演化分析中学习信息,而且还从模板中学习结构信息。这种结合使用数据驱动和知识驱动方法的策略有可能提高预测的准确性和鲁棒性。
需要注意的是,模板引导的预测可能对模板的质量和与目标序列的相似度高度敏感,而且不适用于那些无法找到合适模板的蛋白质。此外,这种方法可能需要更复杂的训练步骤和更多的计算资源。
AlphaFold2 architecture
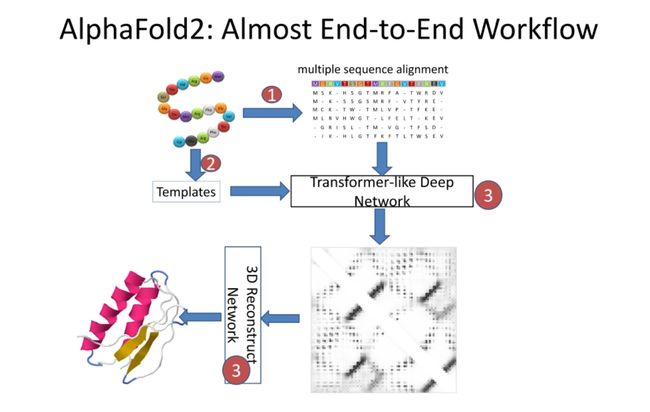
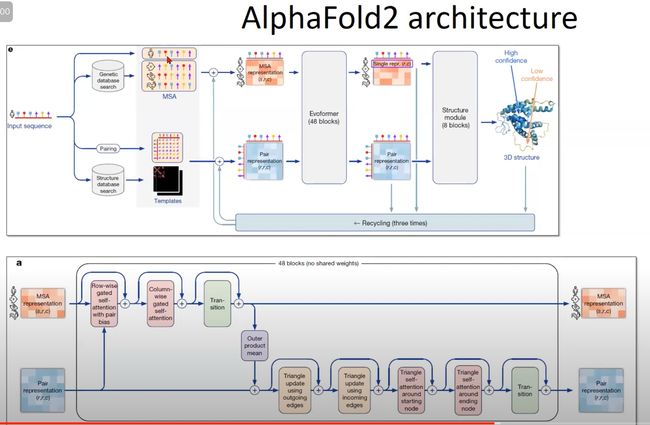
AlphaFold2是一种革新性的蛋白质结构预测方法,由DeepMind公司开发。它在2020年的蛋白质结构预测竞赛(CASP14)中表现出色,标志着蛋白质折叠问题的一个重大突破。
这种方法是端到端的,这意味着它接受一系列氨基酸(即蛋白质序列)作为输入,并输出预测的蛋白质三维结构。这与许多早期的方法不同,这些方法通常需要一系列中间步骤和手动调整。
在AlphaFold2的架构中,首先执行多序列比对,并搜索模板。然后,这些信息被喂入一个类似于Transformer的深度网络中。这个网络包括多个模块,如Evotransformer模块和Structure模块,这些模块通过大量的注意力机制和残差连接来处理序列和模板信息。
网络的输出是一个三维表示,描述了每个氨基酸在空间中的预期位置。这个表示随后被用来重构蛋白质的三维结构。
AlphaFold2的一大创新是引入了模板和多序列比对信息,这些信息通过一个强大的Transformer-like网络进行处理。这允许网络学习复杂的序列-结构关系,并提高预测的准确性。
然而,虽然AlphaFold2取得了显著的成就,但仍有一些挑战。例如,对大蛋白质的预测,以及对蛋白质动态和蛋白质-蛋白质相互作用的预测,仍然是困难的问题。
图神经网络的关键就是学习各个残基之间的关系,完成某些三维结构中每个残基的嵌入,预测最后的三维结构
在蛋白质结构预测的情况下,GNN可以被用来模拟蛋白质折叠的过程,其中节点代表氨基酸,边代表氨基酸之间的相互作用。GNN可以捕获蛋白质的局部和全局结构特征,以及氨基酸之间的复杂相互作用。
例如,可以使用GNN来预测氨基酸之间的距离,这是蛋白质三维结构预测的一个关键步骤。GNN可以通过在蛋白质图中传播信息,来捕获氨基酸之间的长距离依赖性。
补充:跟李沐精读论文
李沐老师b站:AlphaFold 2 论文精读【论文精读】
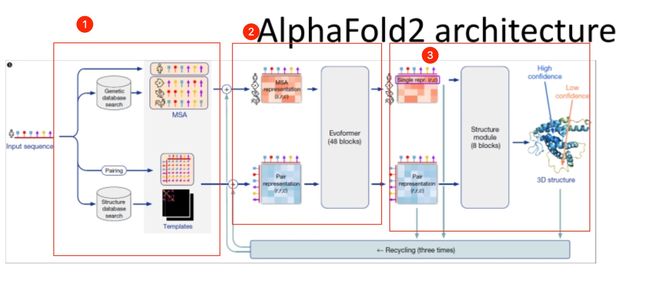
三个部分,第一个部分是抽取特征,第二个是encode,第三个是decode
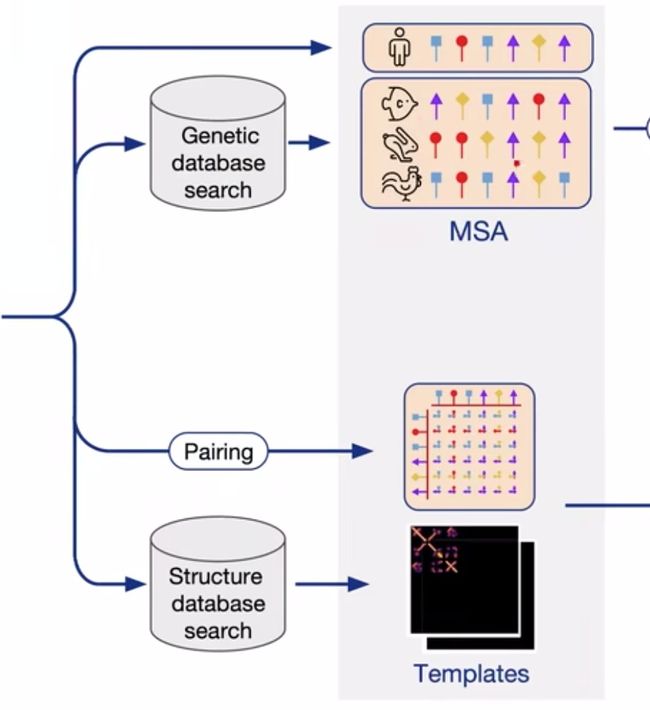
- 对于第一部分,总共是以下几种输入
- 直接导入该序列
- MSA:在基因库中搜索,找相似蛋白质序 列,然后形成一个MSA(Multi sequence alignment),也叫做多序列比对。(MSA的作用是为了提取出一个蛋白质序列在多物种中的共进化信息)
- 氨基酸之间的关系,我们知道蛋白质能卷起来,就是氨基酸之间的关系,所以有一个输入的数据,是存储该蛋白质序列中,氨基酸之间的关系的(这里不是氨基酸间的空间距离,因为我们还不知道,是一些其他方面的特征)
- 最后还有一些额外特征:在结构数据库里搜,因为我们已经知道一些蛋白质的结构,然后在其中搜索得到氨基酸之间的空间距离之类的信息,得到很多模版
- 所以抽取特征主要得到两大类特征:第一类是不同序列之间的特征,第二类是氨基酸之间的特征。这两类特征再拼上一些别的东西,就可以输入编码器了
-
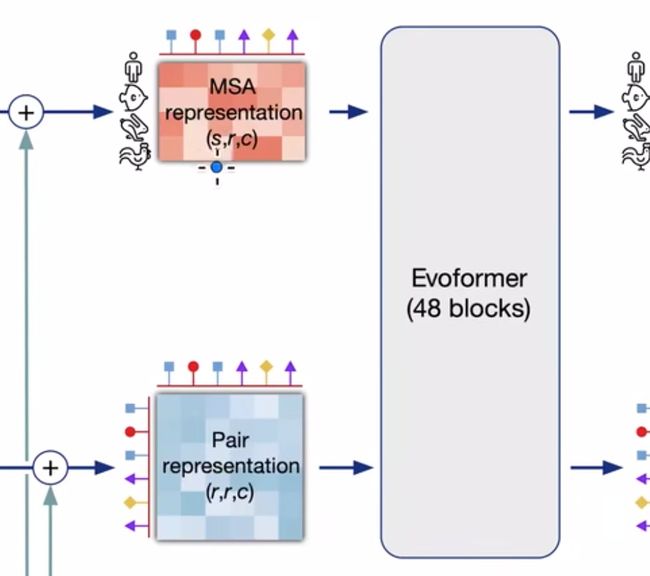
对于第二部分,是输入两个三维的张量
- MSA:大小为(s,r,c)
- s表示有s个蛋白质,第1个是我们要预测的人类的蛋白质,后面s-1个是从数据库中匹配来的蛋白质。
- r表示蛋白质中有r个氨基酸(多序列比对的结果,会用_补齐空缺,最后的结果应该是同长的)
- c表示每个氨基酸表示成长为c的向量(对于image就是每个像素的通道数,对于句子来说就是每个词的嵌入长度)
- Pair氨基酸对:大小为(r,r,c)
- r,r就是氨基酸个数,c就是用长为c的向量来表示一个氨基酸的特征
- MSA:大小为(s,r,c)
-
然后将两个输入进Evoformer(可以看做transformer的变种)
- 与transformer有两个不一样的地方:1.不再是一个序列的关系(比如句子),现在是一个二维之间的关系(不同蛋白质序列、同一氨基酸位点)2. 输入的是两个不同的张量,我们要融合起来
- 其他大部分就一样了。猜到可能用了transformer蛋白质3D结构也是氨基酸相互之间关系造成的,而且序列位置近的和序列位置远的都可能起重要作用,有attention 的味道了
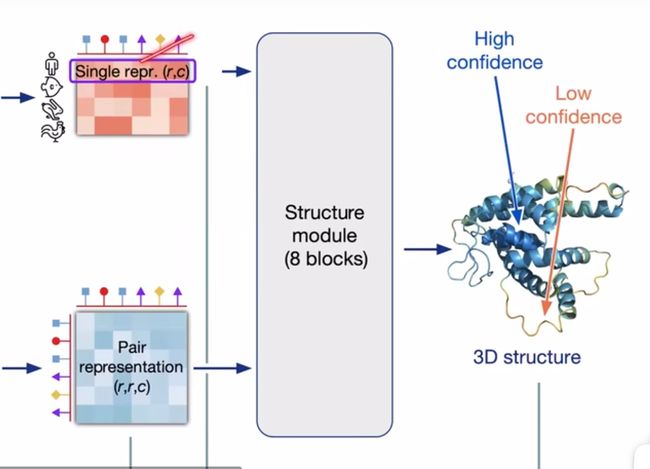
- 然后我们得到了编码器的输出,包括要预测的人类氨基酸的所有的特征表示,还有氨基酸之间的相关信息。根据相关信息来预测每一个氨基酸的位置,最后得到我们的输出
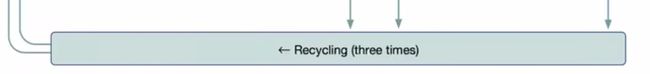
这里还有一个回收机制,将编码器和解码器的输出,又拿回去做了编码器的输入。有点像RNN,可以看做变成了一个四倍更深的一个网络,达到更好的进度。
一些不同:每次复制的权重还是基于前面的,还有做回收的时候梯度是不反传的
暂停来自视频23:05,编码器和解码器的内部结构以后再来补充
摘录了一些小伙伴的b站评论:
问:首先是一个不成熟的小建议:我觉得李老师在做AI for science这类论文的解读时,要是能够对这个science任务做个简要介绍就更好了。比如这个任务是要从什么已知条件得到什么未知结果,以及目前非AI方向是用哪一些方法来完成这个任务。这应该能有助于我们更好地理解作者在这篇文章中某些做法的意图。这是我这几天找的资料整理的,生信的小伙伴们看看有没有什么问题:
蛋白质的三级结构是由一级结构决定的,每种蛋白质都有自己特定的氨基酸排列顺序,从而构成其固有的独特的三级结构。蛋白三维结构预测就是指输入一段蛋白序列(一级结构),输出蛋白所有原子的三维空间坐标。当前对蛋白质三位结构进行预测的方法,除了文中提到的Cryo-SEM,还有通过同源蛋白质的同源建模方法。具体步骤如下:首先选择最佳模板3D结构后进行序列比对,第一次的序列比对通常使用BLOcks替换矩阵执行。第二次序列比对(也称为比对校正)用于构建骨干三维结构。然后对无模板区域或者相似性比较低的区域进行loop建模,最高精度可达12~13个残基。接着是侧链重建,通过依赖主链的旋转体库进行构象搜索。接下来应该通过各种质量评估工具对结构进行改进和验证。我想在本文中输入的MSA,正是对应模版的蛋白质序列,而template也就是这些模版蛋白质序列对应的结构信息。
答1:MSA和template并不一样,MSA的序列远比template里面的序列多的多。MSA建模的思想是来自共进化分析,也就是只通过MSA完全不要序列理论上也能得到3D结构;template建模就是您说的同源建模的思想。但是AF里面保证精度的核心是MSA
答2:MSA 是在seq databse里搜索和查询目标相似的序列来提取共进化信息,也就是在序列层面上获取一些残基之间的接触关系来指导最终的结构预测,template 基于序列直接搜索同源结构,获取的是模板结构在三维空间中残基之间的相对位置信息。MSA 的适用性要更广于template,主要原因在于蛋白质序列的数据库规模要远大于解出结构的蛋白质构成的结构数据库。
5. Computational Drug Development
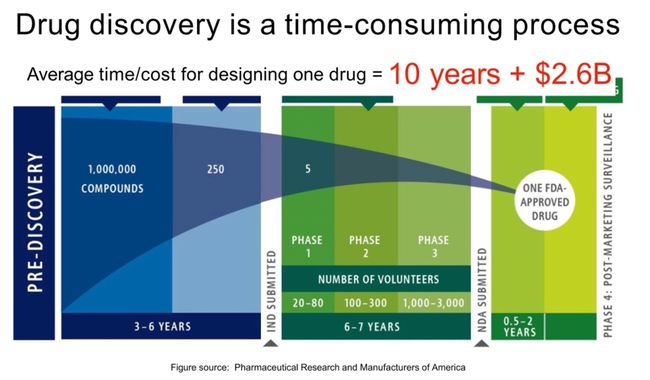
从数百万种化合物中,找到我们做实验用的几种化合物。设计一个药物的时间和金钱成本是巨大的
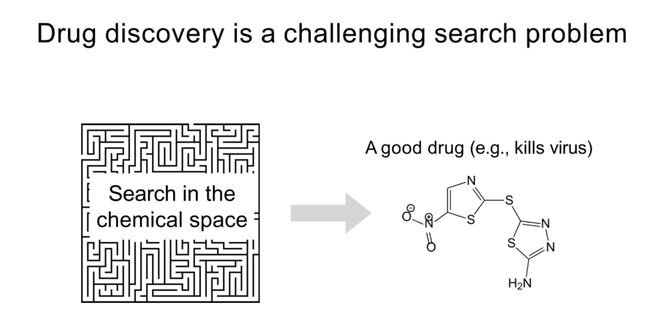
药物发现的过程包括几个阶段:
- 目标识别和验证:在这个阶段,科学家们会识别在疾病过程中发挥关键作用的分子或通路,并验证其作为药物靶点的潜力。
- 筛选和优化引物:在此阶段,大量的化学物质库被筛查,以识别与目标互动的"命中"化合物。这些"命中"化合物然后被优化成"引物"化合物 - 有可能成为药物,能对目标产生期望的影响,且安全、可以有效生产。
- 临床前测试:在实验室和动物模型中对引物化合物进行测试,以评估其在人体内测试之前的安全性和有效性。
- 临床试验:临床试验涉及在人体内测试药物。它们通常分为三个阶段。第一阶段的试验涉及到一小组健康的志愿者,目的是评估安全性和剂量。第二阶段的试验涉及到更大的患者群体,目的是评估效果和副作用。第三阶段的试验涉及到大群患者,目的是确认效果,监控副作用,并将药物与常用的治疗方法进行比较。
- 监管批准:如果临床试验的结果是积极的,药品公司可以申请监管批准以销售药物。
Computational drug discovery: three schemes

计算机辅助药物发现有三种主要的策略:
- 功能空间模拟:传统方法,这种方法利用计算机模拟,例如解决薛定谔方程,来理解和预测分子的物理和化学性质,如氧化还原电位,溶解度和毒性。这种理论的方法通常需要高计算能力。
- 虚拟筛选:虚拟筛选是一种计算策略,通过筛选大量的化合物库,识别出与目标蛋白质或其他生物大分子最有可能相互作用的候选化合物。高通量虚拟筛选一般有多个过滤阶段,包括分子对接、ADMET(药代动力学)性质预测等,以确定最有可能的药物候选物。
- 全新药物设计:这是一个相对较新的领域,其使用优化算法,演化策略和生成模型(如变分自编码器(VAE)、生成对抗网络(GAN)和强化学习(RL))在化学空间中搜索和设计新的候选药物分子。全新药物设计的目标是生成在化学空间中可能尚未被探索的新型化合物,这些化合物可能具有优良的药理活性和适当的药代动力学特性。
6. Virtual drug screening
Virtual screening
虚拟筛选
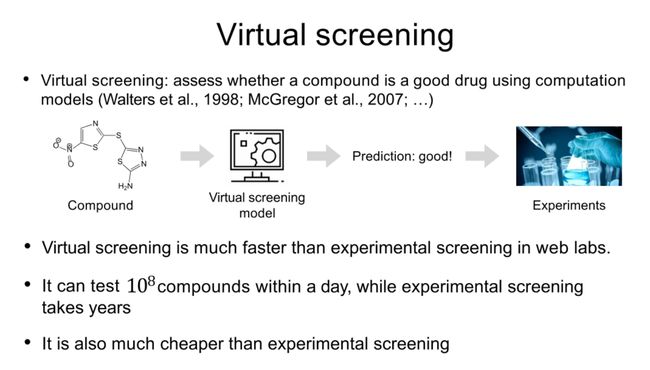
虚拟筛选是使用计算模型来评估一个化合物是否是好的药物(Walters等人,1998; McGregor等人,2007)。
虚拟筛选模型 --> 预测:好的! --> 化合物 --> 实验
虚拟筛选比实验室的实验筛选要快得多。它可以在一天内测试10的9次方种化合物,而实验筛选需要花费数年时间。同时,虚拟筛选也比实验筛选更便宜
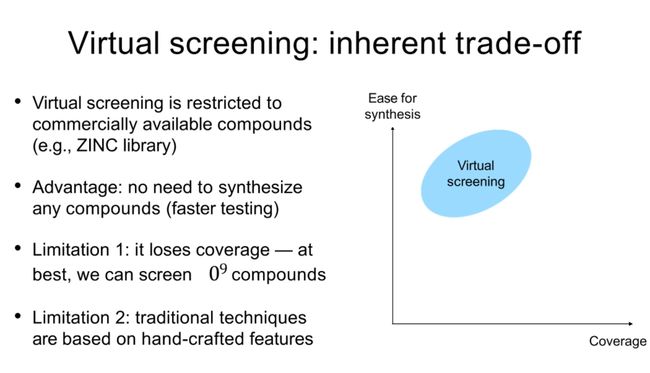
虚拟筛选是一种内在的权衡
- 虚拟筛选限制在商业上可获取的化合物(例如,ZINC库)。
- 优点是不需要合成任何化合物(测试更快)。
- 限制1:它失去了覆盖面,在最好的情况下,我们可以筛选10的9次方种化合物。
- 限制2:传统技术基于人工设计的特性
Part 1: antibiotic discovery 抗生素
抗生素的滥用导致了耐药性的细菌的生长。1987年之后就没有新的抗生素种类的引入了。而因为抗药性,我们需要研发新的抗生素。
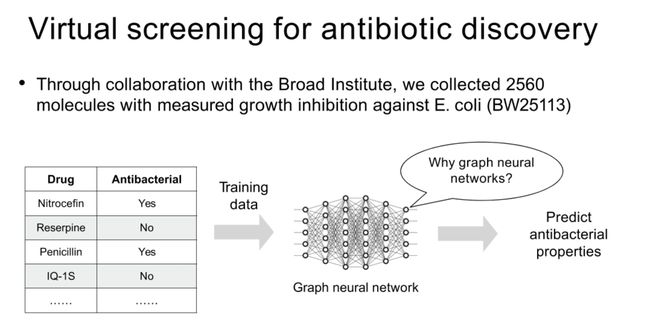
虚拟筛选用于抗生素发现
-
通过与Broad研究所的合作,我们收集了2560种对大肠杆菌(BW25113)的生长抑制测量结果的分子。
-
为什么选择图神经网络?
-
我们有一些药物数据,例如Nitrocefin、Reserpine、Penicillin和IQ-1S,它们是否具有抗菌性质,我们通过训练,然后预测其抗菌性质。
图神经网络可以自动从数据中学习特征表示。
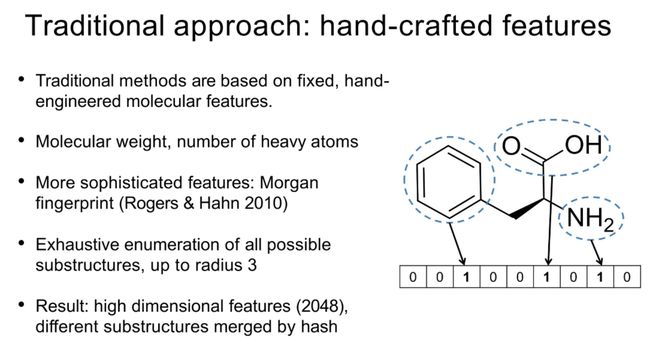
Traditional approach: hand-crafted features
-
传统方法:手工制作的特性
- 传统方法基于固定的、手工工程化的分子特性。这些特性包括分子重量,重原子的数量等。更复杂的特性如Morgan指纹(Rogers & Hahn 2010),通过枚举所有可能的子结构,直到半径为3。结果是高维特性(2048),不同的子结构通过哈希合并。
-
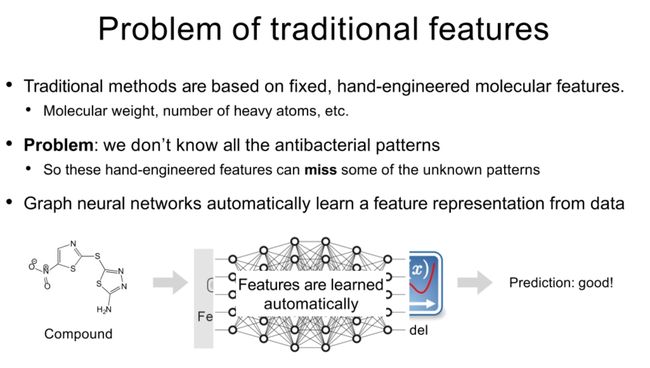
传统特性的问题
- 传统方法基于固定的、手工工程化的分子特性。但是问题在于,我们并不知道所有的抗菌模式,因此这些手工设计的特性可能会错过一些未知的模式。
- 图神经网络可以自动从数据中学习特性表示。特性是自动学习的,例如,给定一个化合物,网络可以自动预测它的特性,从而预测它是否好。
Graph neural network (GNN)

用图来表示化合物分子,edge是键,node是原子,node的属性不止是原子本身,还包括embedding来的(从周围)
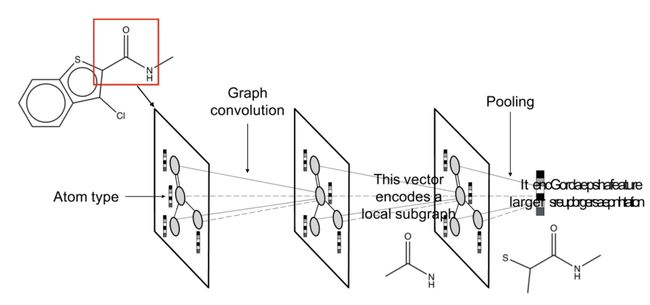
像卷积一样,从原子类型开始,然后根据其邻居的属性来学习,再然后根据邻居的邻居的属性来学习。通过图结构的传播,最终对整个分子的属性进行编码
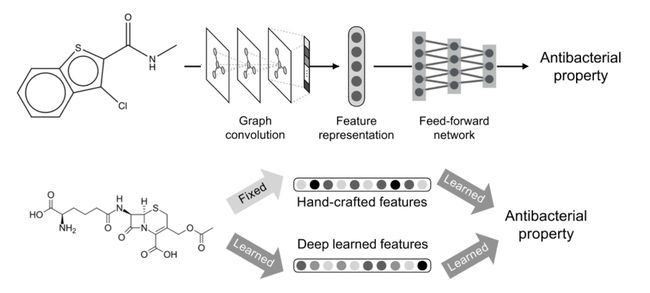
结合手工的特征和深度学习的特征,来判断是否有抗菌性
Use GNN for virtual screening
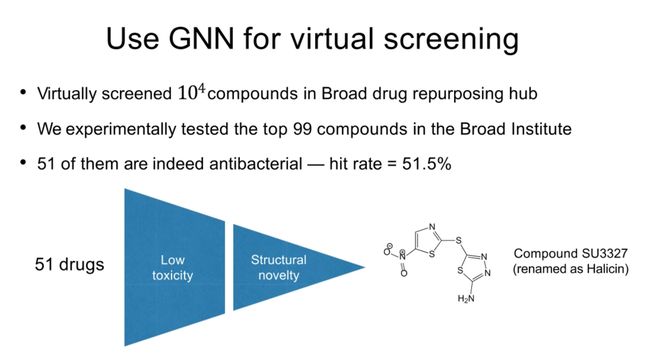
我们在Broad药物再利用中心虚拟筛选了10^6种以上的化合物,并在Broad研究所对排名前99的化合物进行了实验测试。结果发现,其中的51种化合物确实具有抗菌活性,命中率为51.5%。
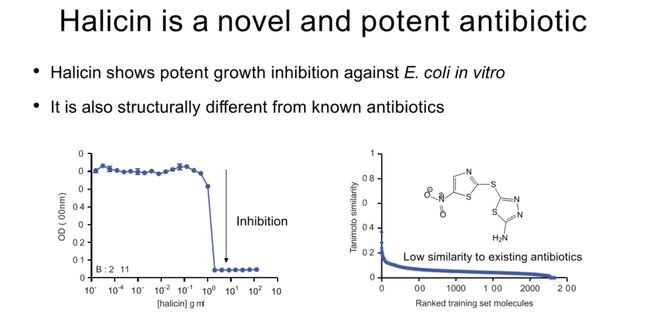
SU3327化合物(重命名为Halicin)是一种新型和强效的抗生素。在体外实验中,Halicin对大肠杆菌表现出强力的生长抑制作用,其结构也与已知的抗生素明显不同,与现有抗生素的相似性低。

Halicin对抗耐药细菌的效力在老鼠身上得到了验证。对抗多重抗药性的鲍曼不动杆菌(A. baumannii)以及抗药性艰难梭菌(C. difficile)在体内都展示出强烈的抑制效果。
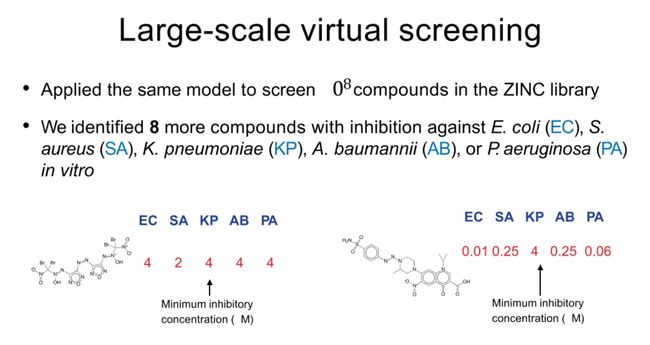
我们还对ZINC库中的10^9种化合物进行了大规模虚拟筛选,成功鉴定出8种对大肠杆菌(EC)和耐药金黄色葡萄球菌(S)具有抑制作用的化合物。

7. De novo drug design
比虚拟筛选更进一步的,创造新药物。我们使用生成模型去尝试。

“从头”药物设计是直接生成具有期望属性的化合物的过程(Moon等人,1991;Clark等人,1995;Schneider & Fechner,2005;)。在这个过程中,我们需要解决反问题:根据期望的属性(例如,氧化还原电位,溶解度,毒性)在功能空间和化学空间中找到一个好的药物。
“从头”药物设计的固有权衡:虚拟筛选仅限于商业化合物(例如,ZINC库)。优点是能有效地探索整个化学空间。但是限制在于我们需要合成新化合物,这可能很难。“从头”药物设计基于手工设计的规则(例如,遗传算法)来探索空间。
设计新药的动机:深度学习可以发现新的抗生素和COVID-19药物。简单的方法是训练一个图神经网络对所有在库中的化合物进行排序。原因是这样可以最大化实验验证的速度。问题在于,像药物的分子数量达到了10^60,我们无法对所有分子进行排序。
为“从头”药物设计进行图生成:我们要学习一个分布,其质量集中在“好”的分子附近。我们可以训练一个生成模型直接生成“好”的分子。这种方法可以有效地探索整个化学空间(10^60种分子)。接下来的问题是如何生成分子图?
Previous solution 1: sequence-based methods
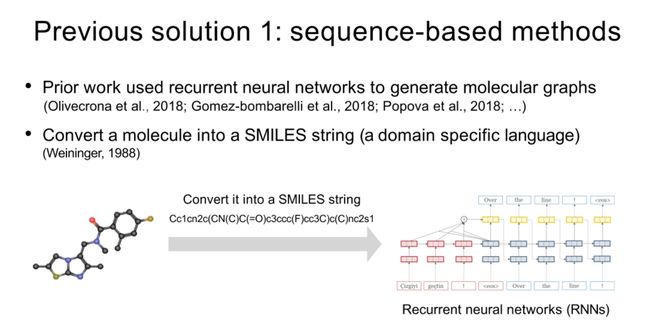
- 这里使用了SMILES字符串来表示化合物
- 先前的工作使用递归神经网络生成分子图表。他们把一个分子转换成一个SMILES字符串(一种特定领域的语言)。
- 但这种基于字符串的表示法是非常脆弱的…几乎相同的两个图表,他们的字符串表示是非常不同的。
- 以前的解决方案:节点逐个生成
- 一个直接的方法:一次生成一个节点的图表(Liu等人,2018)。分子通常是稀疏的:N个节点,O(N)个边。
- 告诉我们就是不要想着单个节点,直接按一个子图去想。
Junction tree variational autoencoder
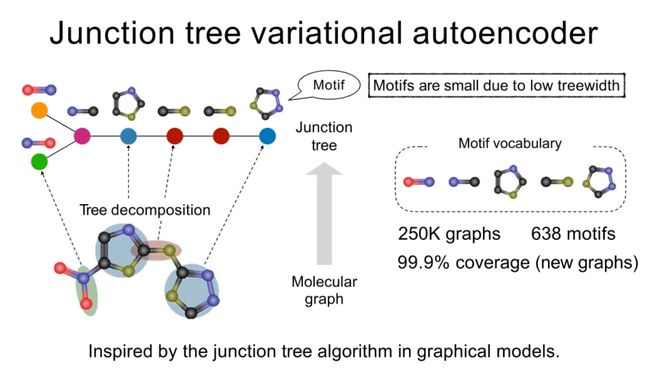
使用motif来分解替代表示。638个motifs涵盖了250k的图
"结点树变分自编码器"的设计灵感来源于图模型中的结点树算法。这种方法考虑了分子图中常见的结构模式(motifs),将复杂的分子结构分解为较小的部分。
这些小部分被称为"motifs",它们因树宽度低而规模较小。我们可以从大量图表(例如,250K图表)中提取出这些模式,形成一个"motif词汇表"。
然后,我们将这些motifs重新组合成新的分子结构。这个过程覆盖了99.9%的新图表,这表明我们可以生成几乎任何类型的分子结构。
在这种方法中,分子图表被表示为其对应的结点树,这使得我们可以有效地对图表进行操作并从数据中自动学习特征表示
用结点树来表示分子图
Details: hierarchical encoder & decoder
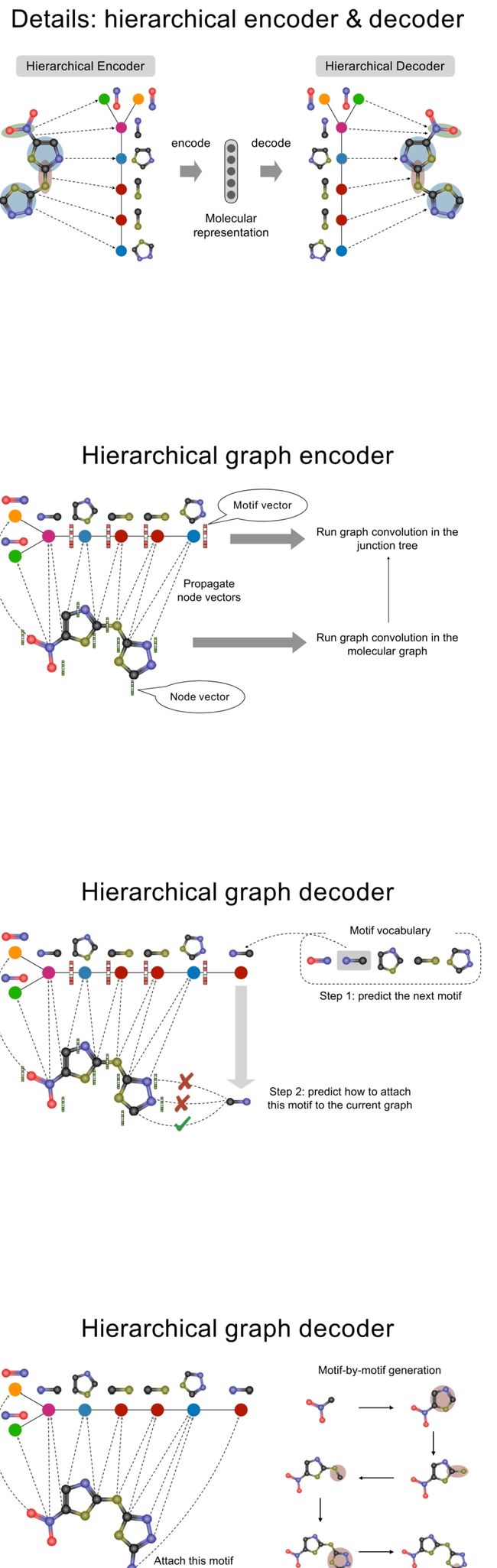
- 分层编码器:
- 首先,使用图卷积运行在分子图表的交叉树(junction tree)上,生成每个motif的向量表示。
- 然后,将这些节点向量传播到分子图的各部分,以在这一级别上运行图卷积。
- 结果是我们得到了一个多层次的、包含从局部到全局信息的分子表示。
- 分层解码器:
- 分层解码器则负责生成新的分子结构。首先,它预测下一个要生成的motif。
- 然后,它预测如何将这个motif附加到当前的图表上。这个过程按照motif-by-motif的方式逐步进行,直到生成完整的分子结构。
这种分层的编码-解码过程允许模型捕获和生成复杂的分子结构,同时保持计算效率。
Motif-by-motif versus node-by-node
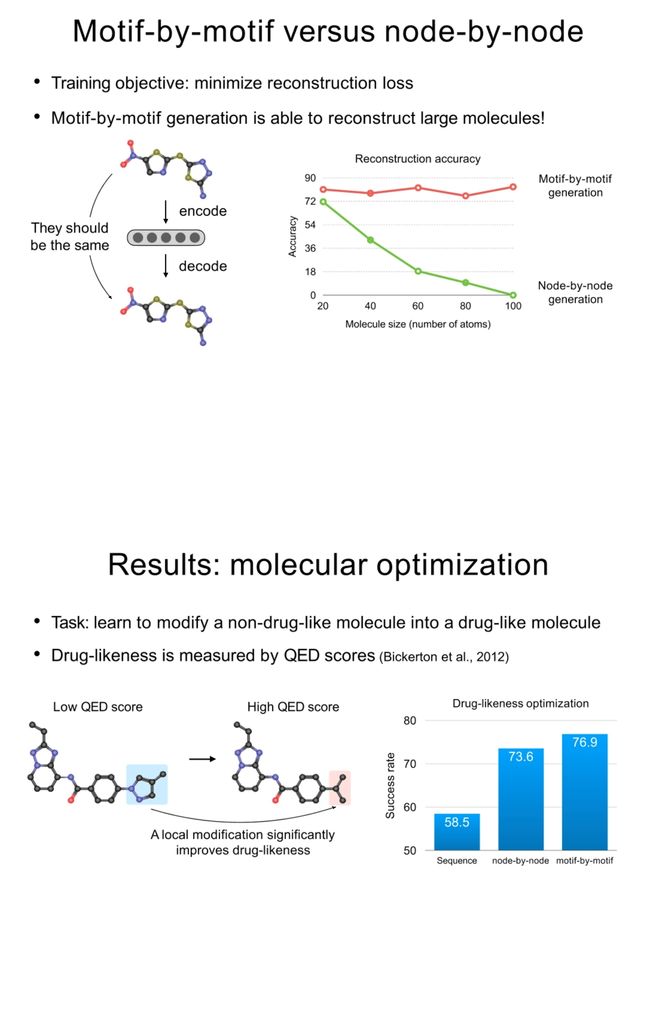
Motif-by-Motif 对比 Node-by-Node
训练目标:最小化重构损失
与逐节点生成相比,Motif-by-Motif(按motif生成)方法能够更好地重构大分子!
图中的重构准确性展示了这种差异。Motif-by-Motif生成的准确性随分子大小(原子数量)的增加而保持稳定,而逐节点生成则在分子大小增加时,准确性显著下降。
结果:分子优化
任务:学习将非药物样的分子修改为药物样的分子。药物样性由QED分数(Bickerton等人,2012年)衡量。
如图所示,Motif-by-Motif生成方法在提高药物样性的成功率上表现得更好,比逐节点和序列方法都要高。所以,这证明了使用Motif-by-Motif的方法,在保持分子结构复杂性的同时,能够有效地优化分子以达到预期的属性。
8. Research Frontiers