Nature Communications | 张阳课题组:端到端深度学习实现高精度RNA结构预测
RNA分子是基因转录的主要执行者,也是细胞运作的隐形功臣。它们在基因表达调控、支架构建以及催化活性等多个生命过程中都扮演着关键角色。虽然RNA如此重要,但由于实验数据的缺乏,准确预测RNA 的三维空间结构仍然是目前计算生物学面临的重大挑战之一。
近日,新加坡国立大学张阳教授团队在《Nature Communications》杂志上发表了题为 “Integrating end-to-end learning with deep geometrical potentials for ab initio RNA structure prediction” 的研究论文。该研究报道了一项用于精确预测RNA的三维结构全新技术DRfold。新加坡国立大学和密歇根大学的李阳博士、张成辛博士和封晨洁博士为该论文的共同第一作者。

图1: 新加坡国立大学张阳团队在《Nature Communication》上的论文
研究背景
就像蛋白质分子一样,RNA分子的生物学功能与其特定的三维构象密切相关;而这种三维构型取决于RNA分子中核苷酸序列的排列顺序。RNA分子的序列和结构,会影响到RNA的稳定性、药物结合、催化活性等多个关键功能。因此,深入了解RNA的三维结构对于疾病研究和药物开发至关重要。然而,传统X射线晶体、冷冻电镜和核磁共振等生物湿实验方法不仅耗时,而且成本高昂,远远无法满足生物学家的结构测定需求。因此,直接基于RNA序列进行结构建模的计算方法成为现代计算生物学一个前沿热点方向。
DRfold采用了一种RNA的粗粒度模型,并将RNA结构的特定原子进行建模,以达到提高计算效率的目的。特别是,研究人员引入了一个与端对端训练并行运行的新颖几何模块设计,同时整合端对端学习和几何势,指导RNA结构的模拟重建。研究结果令人振奋,DRfold方法能够生成比传统自动方法更准确的RNA结构模型。另外,定量的基准测试结果表明,端到端深度学习与几何学相结合,超越了仅基于粗粒度端对端学习或仅基于几何的结构优化方法。
DRfold的执行代码可以在张阳教授团队的实验室网页(https://zhanggroup.org/DRfold/)免费获得。该网页也提供在线服务。使用者只需提供RNA序列,DRfold服务器自动生成RNA三维结构模型。
DRfold算法流程
DRfold流程如图2A所示。首先,通过用户提供待预测的RNA序列,DRfold 预测该序列的二级结构特征;其次,这些信息被送入嵌入层(图2B),它的任务是将输入的RNA序列和二级结构信息转化为计算机可以理解的表示形式。
随后,这些表示形式通过 48 个 RNA transformer 模块(图2C),进行了一系列复杂的转换。这个过程中,深度学习将自动学习如何通过结构模块(图2D)预测RNA的全局结构,包括每个核苷酸的旋转和平移信息(图2G),这些信息将用于后续恢复RNA特定原子(图2F)坐标。
同时,还有另一组神经网络模型(图2E)专门用于预测RNA核苷酸之间的几何结构(图2H)。所学到的全局结构信息和核苷酸间的几何信息将被整合成一个复合势能。这个复合势能描述了RNA分子的稳定性和构象。
随后,计算机使用基于梯度的优化算法,尝试不断调整RNA分子的构象,以找到具有最低能量的最稳定的结构。最终,这个优化后的构象被选定作为DRfold的输出模型。
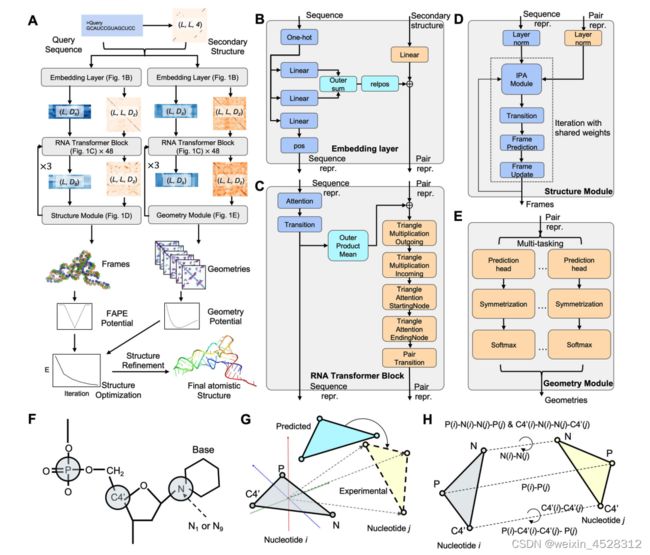
图2. DRfold利用深度学习预测RNA分子结构流程图
实验结果
与其他算法的比较
这项研究首先将DRfold与以前的RNA结构计算机预测方法进行了比较。对比方法包括片段组装方法和从头预测方法,计算结果表明DRfold在多项性能指标上均超越了这些方法(图3)。例如,与其他方法相比,DRfold的模型在RMSD误差和TM-score方面表现出更高的准确性,这表明它可以更准确地预测RNA结构的三维构象。
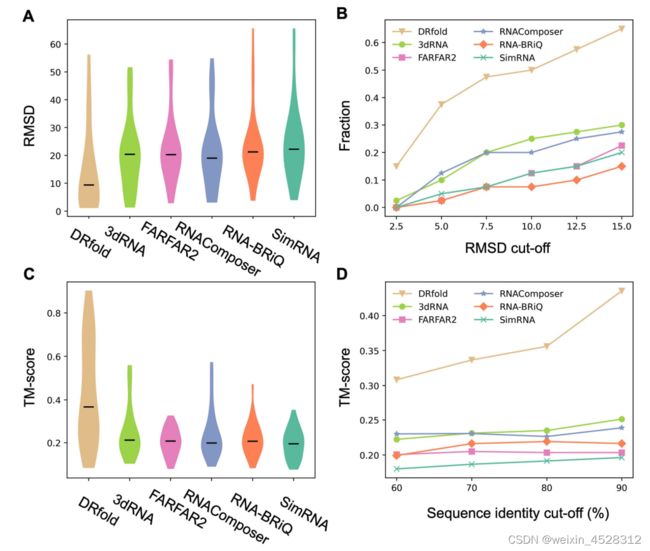
图3. DRfold与其他方法的性能比较
DRfold的性能优势
DRfold的性能优势与研究团队的预期一致。因为现有的自动RNA结构预测方法主要利用基本的经验和统计势能的形式来进行预测,这种形式通常表示为(|)。由于这些方法的力场中参数数量有限,因此无法广泛考虑全局序列条件,通用的势能形式(例如距离或角度)也不能精确地确定RNA结构的复杂拓扑结构。相比之下,DRfold使用的Transformer模块嵌入了大量的权重参数设置,使其能够访问全局序列信息。这意味着DRfold可以更好地捕捉RNA序列中的全局特征,从而提高了结构预测的准确性。此外,DRfold还采用了端到端的损失函数,确保深度学习的预测与正确的整体构象高度一致。这意味着DRfold不仅可以考虑全局序列信息,还可以确保生成的结构与实际的RNA结构紧密匹配。
氢键的作用
在RNA分子中,氢键相互作用在三维结构中起着至关重要的作用,直接影响着RNA的结构和生物功能。准确地预测这些关键的结构模式对于推进我们对RNA生物学的认识至关重要。尽管DRfold没有在其方法中明确纳入特定的碱基配对信息,但它在相关评估指标上的表现都优于其他方法(表1)。
表1. DRfold与其他方法的RNA二级结构恢复性能比较
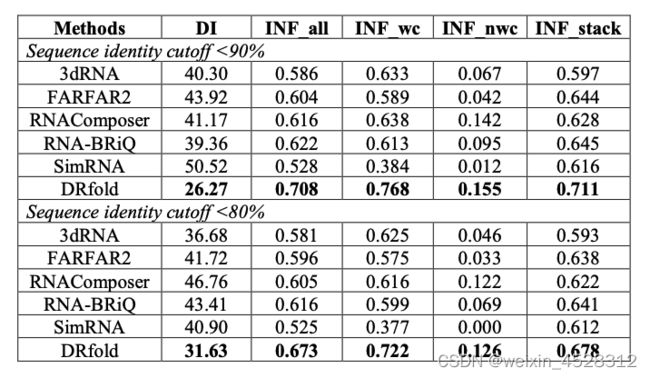
这表明在DRfold的方法中,通过学到的相对位置的帧对齐点误差(FAPE)和几何分布,可能形成了一种隐含的机制,有助于恢复RNA结构内的碱基配对模式。然而,值得注意的是,DRfold 在预测非经典碱基配对模式(INF_nwc)方面的成功率仍然相对较低。为了在这一特定方面提高准确性,研究人员建议开发一个更详细的原子水平学习模型。通过增强非经典配对示例的数据集权重,从而期望提供更准确的预测。
伪结(pseudo-knot)的预测
该项研究的测试集中涉及了40个RNA测试目标,其中15个包含被称为伪结(pseudo-knot)的复杂结构。有趣的是,虽然其他方法难以准确检测到这些伪结,但DRfold成功预测了两个与原生结构中观察到的伪结非常相似的结构(图4)。这突显了DRfold学习和复现RNA分子内复杂碱基间相互作用模式的出色能力。
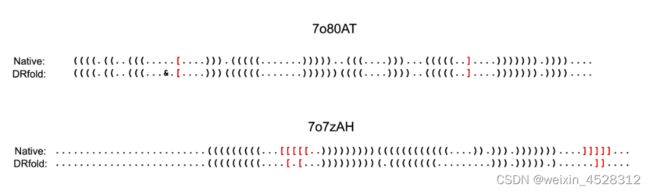
图4. DRfold成功预测两个含有伪结的RNA分子的结构。红色部分表明伪结结构
FAPE势能和几何势能的互补
DRfold的核心创新在于引入了两种互补的势能函数:FAPE势能和几何势能。它们通过两个独立的Transformer网络进行训练,共同构成了RNA结构预测的深度学习势能。
首先,FAPE势能的作用是直接预测核苷酸的旋转矩阵和平移向量,也就是核苷酸构象的坐标信息。通过这种方式,DRfold能够更全面地考虑RNA结构的各种复杂性,从而提高了模型的预测准确性。作者训练了6个独立的端到端模型,每个模型使用不同的参数初始化。令人惊奇的是,即使在没有进行后续处理的情况下,每个单独的端到端模型已经显著优于所有相关的比对算法。这意味着FAPE势能本身已经为RNA结构预测带来了很大的提升。
其次,几何势能使用了一系列几何项,用于表示核苷酸之间的几何关系,包括距离和扭转角。几何势能的引入进一步改善了DRfold的性能。研究人员发现,将几何势能叠加到端到端势能之上,虽然改进不大,但在TM-score上带来了一致的提升。这表明几何势能主要对RNA结构的远距离核苷酸关系起到了积极的作用。
总的来说,DRfold通过端到端学习策略和几何势能的优势互补,让RNA结构预测变得更为准确和全面。消融实验结果如图 5 所示,拿掉任何一个分量,都会降低DRfold的预测精度。
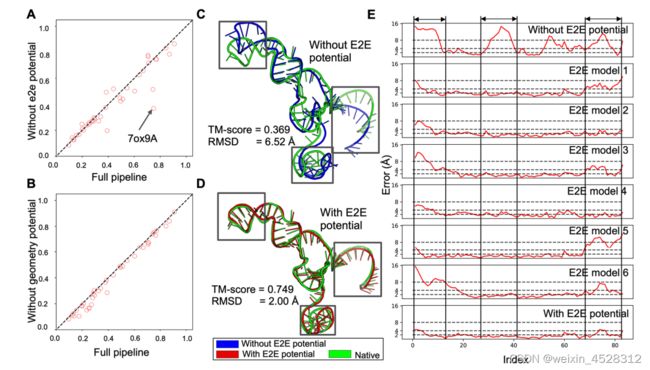
图 5. DRfold中端到端势能和几何势能的消融(ablation)实验结果
单序列vs多序列比对
最近,RNA结构预测领域出现了好几种深度学习模型。这些深度学习方法根据其训练所使用的输入特征可分为单一序列方法和多序列比对(MSA)方法。MSA方法通常可以从多序列比对中获得协同进化信息,因此在整体结构预测方面性能更优。但在单一序列方法中,DRfold表现出色,显著优于其他单一序列方法,而且与大多数MSA方法的性能上相当,差异并不具有统计学上的显著性。
端到端vs几何势
从方法论的角度来看,这些深度学习方法可以分为端到端方法和基于几何的方法。而DRfold则通过势能函数集成将这两种方法结合在一起。这使得DRfold在其方法流程的扩展上更具灵活性。例如,研究人员将DeepFoldRNA的几何预测用于构建混合几何势能,并替换了DRfold中的默认几何势能。这种结合使DRfold获得了更精确的空间约束,有助于更好地引导结构的组装和优化过程。
二级结构预测的作用
因为DRfold是基于单序列的结构预测,二级结构的引导显得特别重要。就像图 6 的结果所示,拿掉二级结构会显著的降低DRfold的模型预测精度。

图 6,二级结构对DRfold模型精度的影响
CASP双盲测试
早期版本的DRfold程序参与了CASP15的RNA结构预测实验,该程序被标识为“rDP”组。尽管只有12个测试目标,仍为评估DRfold的性能提供了宝贵的信息。就RMSD而言,使用惩罚阈值为-2.0和0.0时,DRfold分别排名第5和第6。在使用TM-score评估时,相同的惩罚阈值下,排名分别为第6和第9。然而,DRfold与排名前4的方法(如AIchemy_RNA2和GeneSilico等)之间存在一定的性能差距,这些方法依赖于丰富的RNA结构领域专业知识和人工专业技能。
相比之下,DRfold完全自动运行,且仅利用单一的序列信息进行预测。尽管存在这一差距,CASP15的结果表明,DRfold的基于单一序列的方法在RNA结构预测领域具有很大的潜力。它的性能与一些依赖于额外信息源(如模板、多序列比对或预训练核苷酸序列模型)的方法相媲美。这强调了DRfold在未来版本中通过复合势能函数整合多种信息源来进一步改进的潜力。
结论与展望
DRfold的成功得益于其深度学习模型,这一方法在现有的RNA结构预测中鲜有涉足。DRfold采用的端到端模型已被证明非常有效,再通过整合几何约束,可以进一步提高结构模型的准确性。此外,从物理学角度生成的二级结构特征也对帮助网络学习和生成准确的碱基配对和局部结构组装起到了积极作用。
然而,与蛋白质结构预测相比,RNA结构预测的整体精度仍然有限。这部分原因在于RNA结构可用于训练的实验数据相对较少。此外,为了改进对没有足够同源序列的RNA的建模,DRfold目前只使用单一序列进行训练。进一步使用多序列比对、结构模板和RNA物理知识等额外信息,将显著其结构预测的准确性。
总的来说,DRfold在人工智能RNA结构预测问题上做了一些非常有意义的探索。随着深度学习技术和RNA数据的不断发展,以DRfold为代表的人工智能算法对RNA三维结构的测定将会起着越来越重要作用。
参考文献:
Y Li, C Zhang, C Feng, R Pearce, PL Freddolino, Y Zhang. Integrating end-to-end learning with deep geometrical potentials for ab initio RNA structure prediction. Nature Communications 14: 5745 (2023).
https://www.nature.com/articles/s41467-023-41303-9