原来基因功能富集分析这么简单
本期主要介绍了基因功能富集分析的一些基础概念,同时介绍如何使用DAVID在线分析工具对基因进行GO/KEGG功能富集分析。
什么是基因功能富集分析?
基因功能富集分析,指的是通过借助各类数据库对所获得的众多基因进行统计分析与基因功能分类,从而挖掘在数据库中与我们所研究的生物学问题具有显著相关性的基因功能类别。但需注意的是不同的基因组合在不同生物背景下可能具有不同的生物学功能。因此,需要根据实际情况对相应的基因集进行筛选,并与所研究对象相关的功能变化进行联系,从而将众多差异基因进行归类,并最终聚焦于所研究的生物学问题相关关键差异基因上,从而为后续实验验证提供方向与依据。总而言之,基因功能富集分析的本质就是聚类分析,是用来解读一组基因背后所代表的生物学知识,揭示其在细胞内或外所扮演的角色。
为什么要进行功能富集分析?
随着高通量测序技术的发展,生物学研究领域进入了组学时代。然而,组学测序的庞大数据量,使得研究者望而却步。而对这些数据信息进行有效的提取与分析也成为众多研究者所关注的重点方向。以转录组测序(RNA-seq)为例,测序结果往往会得到一系列差异表达基因,但研究者如何将这些基因与所研究的生物学问题及潜在的调控机制进行联系就成为了关键问题。因此,研究者可通过多个功能注释数据库对基因进行功能富集分析,将这一系列基因集分成不同的功能类别,从中寻找在生物学过程中起关键作用的生物学通路,从而揭示和理解这些生物学过程的基本及潜在的分子机制。事实上,分子水平的落脚点是在基因水平上,但是基因的种类有很多,而理解这些基因所代表的生物学意义的最佳途径就是基因功能富集分析。
依据富集分析过程中基因选择、注释数据库的不同,常用的富集分析可以分为以下几种:GO 功能富集、KEGG 通路富集、GSEA基因集富集等。
GO 功能富集分析
基因本体论(Gene Ontology,GO)数据库是GO联合会在2000年构建的一个数据库,旨在建立一个适用于各种物种的、对基因和蛋白功能进行限定和描述的,并能随着研究不断深入而更新的语义词汇标准。GO注释涵盖了分子生物学功能(molecular function,MF)、生物学过程(biological process,BP)和细胞学组分(cellular components,CC)三个方面,通过这三个功能大类,对一个基因的功能进行多方面的限定和描述。GO注释且是目前应用最广泛的基因注释体系之一。GO知识库是世界上关于基因功能的最大信息源。这一知识既是人类可读的,也是机器可读的,是生物医学研究中大规模分子生物学和遗传学实验的计算分析基础。
分子功能(molecular function,MF):
是描述基因在分子生物学层面的功能或职能,比如催化活性、转运活性、结合活性等。分子功能大部分指的是单个基因产物的功能,还有一小部分是此基因产物形成的复合物的功能。
生物学过程(biological process,BP):
是描述该基因参与了哪些生物学过程,比如参与转录调控、参与rRNA的加工、DNA的复制、细胞生长和维持、信号传导和各类因子运输等过程。生物学过程是由分子功能有序地组成的,具有多个步骤的一个过程。一个生物学途径并不是完全和一条生物学通路相等。因此,GO并不涉及到通路中复杂的机制调控过程。
细胞成分(Cellular component,CC):
描述基因(产物)在细胞存在的位置,比定位在细胞质、细胞核、细胞器、线粒体膜或基质中。又或者在某些基因产物中,比如蛋白酶体等。
GENE ONTOLOGY RESOURCE:http://geneontology.org/
KEGG 通路富集
京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes, KEGG)是系统分析基因功能、基因组信息的数据库,整合了基因组学、生物化学及系统功能组学等方面的数据信息,包括代谢通路(KEGG PATHWAY)、药物(KEGG DRUG)、疾病(KEGG DISEASE)、功能模型(KEGG MODULE)、基因序列(KEGG GENES)及基因组(KEGG GENOME)等。KO(KEGG ORTHOLOG)系统将各个KEGG注释系统联系在一起。KEGG 已建立了一套完整KO注释的系统,可完成新测序物种的基因组或转录组的功能注释。KEGG有助于研究者把基因及表达信息作为一个整体进行研究。
KEGG:https://www.kegg.jp/
GSEA富集分析:
基因集富集分析(Gene Set Enrichment Analysis, GSEA)通常是分析一组基因在某个功能节点上是否相比于随机水平过于出现(over-presentation)的情况。GSEA分析纳入了所有基因,可将一些微弱但不显著的效应都考虑在内。GSEA分析无需做差异分析,直接可拿表达量信息寻找和性状相关的通路/功能基因集合,这样不需筛选的情况可保留一些关键信息,进而找到那些差异不明显但基因差异趋势一致的功能基因集。
接下几期我将主要介绍使用DAVID在线分析工具、R clusterProfiler包等,对基因进行GO、KEGG功能富集分析及相应的可视化。
DAVID在线分析工具对基因进行GO/KEGG功能富集分析
Step 1-2
首先打开DAVID官网:DAVID Functional Annotation Bioinformatics Microarray Analysis,点击“Function Annotation”。
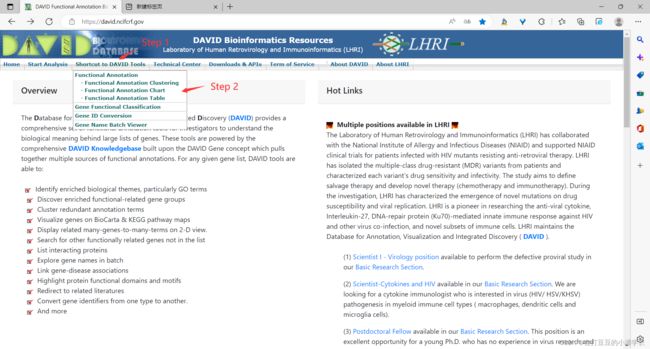
Step 3
导入数据:(1)直接粘贴到“Paste a list”;(2)在“Choose From a File”中直接导入文件,支持txt格式。
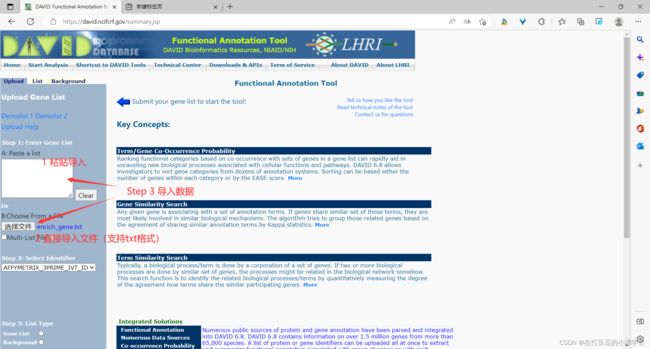 Step 4
Step 4
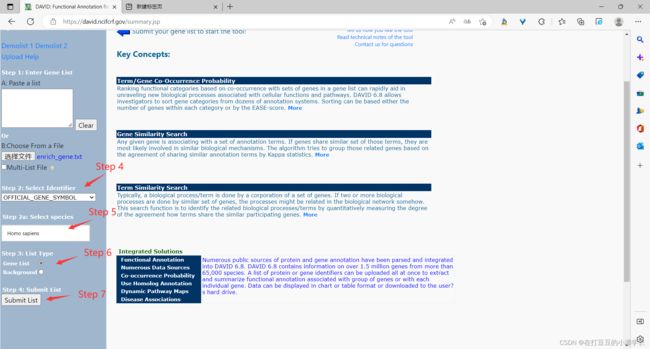
在“Select Identifier”中选择你的基因类型,我上传的为基因名(Gene Symbol),所以我选择“OFFICIAL_GENE_SYMBOL”。(这一步主要根据你导入的数据类型进行选择)
Step 5
在“Select species”中选择你所研究的物种,我这边研究的人,所以我选择“Homo sapiens”。
Step 6
在“List Type”中选择输入的列表的类型,我这边输入的是所研究的基因,所以我选择“Gene List”。
Step 7
点击“Submit List”运行
Step 8
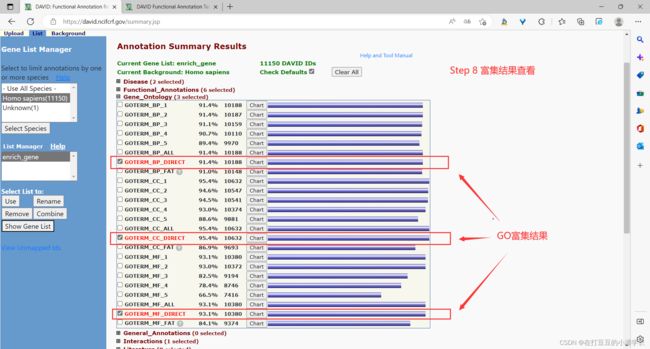
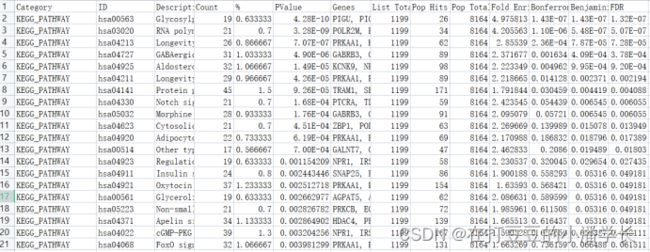
查看数据富集分析结果

Step 9
富集分析结果导出(复制粘贴至Excel)

DAVID所获得富集结果主要由这几列数据构成:Category、Term(GO语义)、Count(基因数)、%(基因比例)、P-Value(P值)、Genes(基因名)、List Total、Pop Hits、Pop Total、Fold Enrichment、Bonferroni(多重检验校正)、Benjamini(多重检验校正)和FDR(校正后的P值)。
 参考文献
参考文献
[1] Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216-221, doi:10.1093/nar/gkac194 (2022).
好了本次分享就到这里,下期将分享对这些功能富集结果进行可视化的方法,敬请期待。

关注“在打豆豆的小潘学长”公众号,收获更多干货。