The pipeline of RNA-seq(菜鸟教学)
一、准备工作
1.1 设置好专门的用于RNA分析的环境
在SRA中下载目标序列,使用wget指令
使用conda激活rna分析环境
1.2 工作目录管理
## 示例如下:
├── database # 数据库存放目录,包括参考基因组,注释文件,公共数据库等
├── project # 项目分析目录
└── Human-16-Asthma-Trans #具体项目
├── data # 数据存放目录
│ ├── cleandata # 过滤后的数据
│ ├── trim_galore # trim_galore过滤
│ └── fastp # fastp过滤
│ └── rawdata # 原始数据
├── Mapping # 比对目录
│ ├── Hisat2 # Hisat比对
│ └── Subjunc # subjunc比对
└── Expression # 定量
├── featureCounts # featureCounts
└── Salmon # salmon定量
# 进入到个人目录
cd ~
## 1.建立数据库目录:在数据库下建立参考基因组数据库,注意命名习惯:参考基因组版本信息
mkdir -p database/genome/GRCh38_release104
## 2.建立项目分析目录
mkdir project
cd project
mkdir Human-16-Asthma-Trans # 注意项目命名习惯:物种-样本数-疾病-分析流程
cd Human-16-Asthma-Trans
# 建立数据存放目录
mkdir -p data/rawdata data/cleandata/trim_galore data/cleandata/fastp
# 建立比对目录
mkdir -p Mapping/Hisat2 Mapping/Subjunc
# 建立定量目录
mkdir -p Expression/featureCounts Expression/Salmon
#查看目录
tree二、原始数据质量评估
2.1 数据质量评估
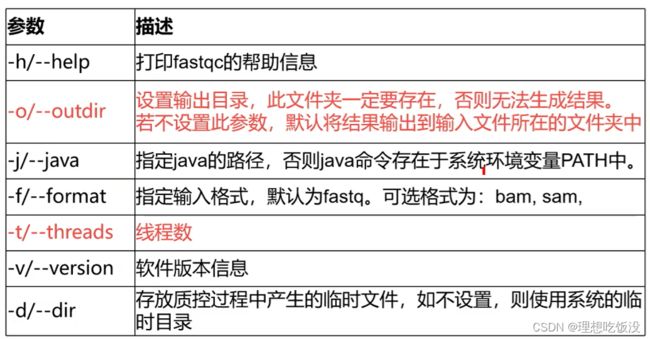
FastQC软件可以对fastq格式的原始数据进行质量统计,评估测序结果,为下一步修建过滤提供参考。
常见参数如下:
# 激活conda环境
conda activate rna
# 连接数据到自己的文件夹
cd $HOME/project/Human-16-Asthma-Trans/data/rawdata
ln -s /teach/t_rna/data/airway/fastq_raw25000/*gz ./
# 使用FastQC软件对单个fastq文件进行质量评估,结果输出到qc/文件夹下
fastqc -t 6 -o ./ SRR1039510_1.fastq.gz
# 多个数据质控
fastqc -t 6 -o ./ SRR*.fastq.gz
# 使用MultiQc整合FastQC结果
multiqc *.zip脚本后台运行: nohup &,两种方式
nohup : no hang up ,退出终端不会影响程序的运行。
&: 后台运行
#plan 1
#直接在命令前后加上nohup &,适用于较短的命令,方便快捷
nohup fastqc -t 6 -o ./ SRR*.fastq.gz &#plan 2
#将命令写入sh脚本,使用nohup &运行sh脚本,适用于比较长和复杂的命令
nohup sh qc.sh > qc.sh.log &三、数据过滤
测序得到的原始序列含有接头序列或低质量序列,为了保证信息分析的准确性,需要对原始数据进行质量控制,得到高质量序列(即Clean Reads),原始序列质量控制的标准为:
(1)去除含接头的reads;
(2)过滤去除低质量值数据,确保数据质量;
(3)去除含有N(无法确定碱基信息)的比例大于5%的reads;
3.1 trim_galore过滤
软件1名称:trim_galore
常用参数:

# 激活小环境
conda activate rna
# 新建文件夹trim_galore
cd $HOME/project/Human-16-Asthma-Trans/data/cleandata
mkdir trim_galore
cd trim_galore
# 单个样本,使用tab键补全路径
trim_galore --phred33 -q 20 --length 36 --max_n 3 --stringency 3 --fastqc -o ./ --paired ../../rawdata/SRR1039510_1.fastq.gz ../../rawdata/SRR1039510_2.fastq.gz
# 先生成一个变量,为样本ID
ls $HOME/project/Human-16-Asthma-Trans/data/rawdata/*_1.fastq.gz | awk -F'/' '{print $NF}' | cut -d'_' -f1 >ID
# 多个样本 vim trim_galore.sh,以下为sh的内容
rawdata=$HOME/project/Human-16-Asthma-Trans/data/rawdata
cleandata=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
cat ID | while read id
do
trim_galore --phred33 -q 20 --length 36 --max_n 3 --stringency 3 --fastqc --paired -o ${cleandata} ${rawdata}/${id}_1.fastq.gz ${rawdata}/${id}_2.fastq.gz
done
# 提交任务到后台
nohup sh trim_galore.sh >trim_galore.log &3.2 fastp过滤
软件2名称:fastp
常用参数:

d $HOME/project/Human-16-Asthma-Trans/data/cleandata
mkdir fastp
cd fastp
# 定义文件夹
cleandata=$HOME/project/Human-16-Asthma-Trans/data/cleandata/fastp/
rawdata=$HOME/project/Human-16-Asthma-Trans/data/rawdata/
# 单个样本
fastp -i $rawdata/SRR1039510_1.fastq.gz \
-I $rawdata/SRR1039510_2.fastq.gz \
-o $cleandata/SRR1039510_1.fastp.fq.gz \
-O $cleandata/SRR1039510_2.fastp.fq.gz \
-h $cleandata/SRR1039510.html \
-j $cleandata/SRR1039510.json \
-l 36 -q 20 --compression=6 -R $cleandata/SRR1039510
# 多个样本
cat ../trim_galore/ID | while read id
do
fastp -l 36 -q 20 --compression=6 \
-i ${rawdata}/${id}_1.fastq.gz \
-I ${rawdata}/${id}_2.fastq.gz \
-o ${cleandata}/${id}_1.fastp.fq.gz \
-O ${cleandata}/${id}_2.fastp.fq.gz \
-R ${cleandata}/${id} \
-h ${cleandata}/${id}.fastp.html \
-j ${cleandata}/${id}.fastp.json
done
# 运行fastp脚本
nohup sh fastp.sh >fastp.log &3.3 数据过滤前后变化
1.有一些reads长度发生了变化
2.有一些reads直接被过滤掉了总read数减少
3.大部分reads都满足过滤条件没有变化
# 进入过滤目录
cd $HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
# 原始数据
zcat $rawdata/SRR1039510_1.fastq.gz | paste - - - - > raw.txt
# 过滤后的数据
zcat SRR1039510_1_val_1.fq.gz |paste - - - - > trim.txt
awk '(length($4)<63){print$1}' trim.txt > Seq.ID
head -n 100 Seq.ID > ID100
grep -w -f ID100 trim.txt | awk '{print$1,$4}' > trim.sm
grep -w -f ID100 raw.txt | awk '{print$1,$4}' > raw.sm
paste raw.sm trim.sm | awk '{print$2,$4}' | tr ' ' '\n' |less -S四、参考基因组的下载
4.1 下载平台
UCSC:http://www.genome.ucsc.edu/
4.2 fasta文件的下载
wget分别下载基因组序列和转录组序列
## 参考基因组准备:注意参考基因组版本信息
# 下载,Ensembl:http://asia.ensembl.org/index.html
# http://ftp.ensembl.org/pub/release-104/fasta/homo_sapiens/dna/
# 进入到参考基因组目录
cd $HOME/database/genome/GRCh38_release104
# 下载基因组序列
nohup wget -c http://ftp.ensembl.org/pub/release-104/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz >dna.log &
# 下载转录组
nohup wget -c http://ftp.ensembl.org/pub/release-104/fasta/homo_sapiens/cdna/Homo_sapiens.GRCh38.cdna.all.fa.gz >rna.log &
4.3 参考基因组注释文件的下载
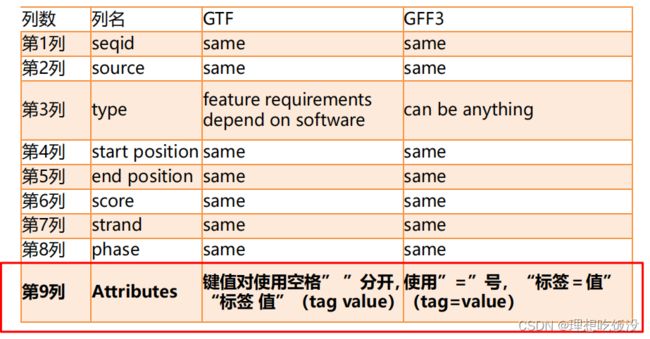
有gtf和gff两种文件格式
gff格式如下:

第九列的详解如下:

两种文件的差异及比较:
# 下载基因组注释文件
nohup wget -c http://ftp.ensembl.org/pub/release-104/gtf/homo_sapiens/Homo_sapiens.GRCh38.104.gtf.gz >gtf.log &
# 解压
nohup gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz Homo_sapiens.GRCh38.cdna.all.fa.gz >unzip.log &五、数据比对
5.1 构建索引
软件:Hisat2 ————转录组数据的比对
命令:hisat2-build 参考基因组fa文件 前缀
hisat2主要参数:

# 进入参考基因组目录
cd $HOME/database/genome/GRCh38_release104
# Hisat2构建索引,构建索引时间比较长,建议携程sh脚本提交后台运行
hisat2-build Homo_sapiens.GRCh38.dna.primary_assembly.fa Homo_sapiens.GRCh38.dna.primary_assembly
# 提交后台
nohup sh index.sh >index.sh.log &5.2 任务串联
三步走:
1. 比对
2. sam转bam (samtools作为工具)
3. bam建索引
单样本
# 进入比对文件夹
cd $HOME/project/Human-16-Asthma-Trans/Mapping/Hisat2
# 输入输出定义文件夹
index=/home/t_rna/database/genome/GRCh38_release95/Homo_sapiens.GRCh38.dna.primary_assembly
inputdir=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore/
outdir=$HOME/project/Human-16-Asthma-Trans/Mapping/Hisat2
# 单个样本比对
hisat2 -p 5 -x ${index} \
-1 ${inputdir}/SRR1039510_1_val_1.fq.gz \
-2 ${inputdir}/SRR1039510_2_val_2.fq.gz \
-S ${outdir}/SRR1039510.Hisat_aln.sam
# sam转bam
samtools sort -@ 6 -o SRR1039510.Hisat_aln.sorted.bam SRR1039510.Hisat_aln.sam
# 对bam建索引
samtools index SRR1039510.Hisat_aln.sorted.bam SRR1039510.Hisat_aln.sorted.bam.bai多样本
# 多个样本批量进行比对,排序,建索引
# Hisat.sh内容: 注意命令中的-,表示占位符,表示|管道符前面的输出。
# vim Hisat.sh
index=/home/t_rna/database/genome/GRCh38_release95/Homo_sapiens.GRCh38.dna.primary_assembly
inputdir=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore/
outdir=$HOME/project/Human-16-Asthma-Trans/Mapping/Hisat2
cat ../../data/cleandata/trim_galore/ID | while read id
do
hisat2 -p 5 -x ${index} -1 ${inputdir}/${id}_1_val_1.fq.gz -2 ${inputdir}/${id}_2_val_2.fq.gz 2>${id}.log | samtools sort -@ 3 -o ${outdir}/${id}.Hisat_aln.sorted.bam - && samtools index ${outdir}/${id}.Hisat_aln.sorted.bam ${outdir}/${id}.Hisat_aln.sorted.bam.bai
done
# 统计比对情况
multiqc -o ./ SRR*log
# 提交后台运行
nohup sh Hisat.sh >Hisat.log &5.3sam/bam格式
比对结果部分
1.每一行表示一个read的比对信息
2.每行包括11给必须字段和1个可选字段,字段之间用制表符分割

sam/bam格式-----FLAG:samtools flags 99
sam/bam格式-----CIGAR:简要比对信息表达式
常用指令如下:

5.4 subjunc比对
常见参数:

构建索引
# 进入参考基因组目录
cd $HOME/database/genome/GRCh38_release104
# subjunc构建索引,构建索引时间比较长,建议携程sh脚本提交后台运行
subread-buildindex -o Homo_sapiens.GRCh38.dna.primary_assembly Homo_sapiens.GRCh38.dna.primary_assembly.fa单样本
# 进入文件夹
cd $HOME/project/Human-16-Asthma-Trans/Mapping/Subjunc
# 输入输出定义文件夹,上课使用的index为教师目录
index=/home/t_rna/database/genome/GRCh38_release95/Homo_sapiens.GRCh38.dna.primary_assembly
inputdir=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
outdir=$HOME/project/Human-16-Asthma-Trans/Mapping/Subjunc
# subjunc单样本比对
subjunc -T 6 -i ${index} \
-r ${inputdir}/SRR1039510_1_val_1.fq.gz \
-R ${inputdir}/SRR1039510_2_val_2.fq.gz \
-o ${outdir}/SRR1039510.Subjunc.bam \
>${outdir}/SRR1039510.Subjunc.log
# 排序以及构建bam索引
samtools sort -@ 6 -o SRR1039510.Subjunc.sorted.bam SRR1039510.Subjunc.bam
# 建索引,.bai结尾的文件为索引
samtools index SRR1039510.Subjunc.sorted.bam SRR1039510.Subjunc.sorted.bam.bai多样本
# vim subjunc.sh
index=/home/t_rna/database/genome/GRCh38_release95/Homo_sapiens.GRCh38.dna.primary_assembly
inputdir=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
outdir=$HOME/project/Human-16-Asthma-Trans/Mapping/Subjunc
cat ../../data/cleandata/trim_galore/ID | while read id
do
subjunc -T 10 -i ${index} -r ${inputdir}/${id}_1_val_1.fq.gz -R ${inputdir}/${id}_2_val_2.fq.gz -o ${outdir}/${id}.Subjunc.bam 1>${outdir}/${id}.Subjunc.log 2>&1 && samtools sort -@ 6 -o ${outdir}/${id}.Subjunc.sorted.bam ${outdir}/${id}.Subjunc.bam && samtools index ${outdir}/${id}.Subjunc.sorted.bam ${outdir}/${id}.Subjunc.sorted.bam.bai
done
# 运行
nohup sh subjunc.sh >subjunc.log &六、表达定量
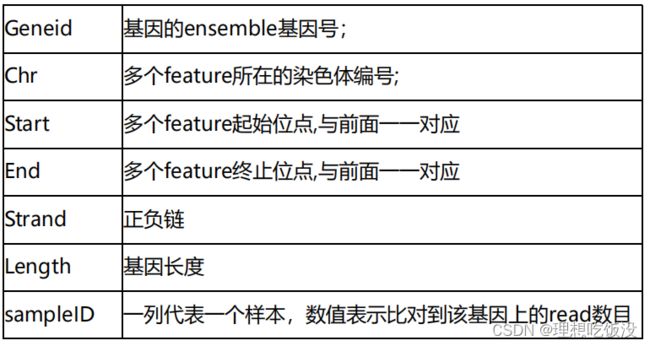
6.1 featurecounts
常用参数如下:

结果解析如下:
cd $HOME/project/Human-16-Asthma-Trans/Expression/featureCounts
## 定义输入输出文件夹
gtf=/home/t_rna/database/genome/GRCh38_release95/Homo_sapiens.GRCh38.95.gtf.gz
inputdir=$HOME/project/Human-16-Asthma-Trans/Mapping/Hisat2/
# featureCounts对bam文件进行计数
featureCounts -T 6 -p -t exon -g gene_id -a $gtf -o all.id.txt $inputdir/*.sorted.bam
# 对定量结果质控
multiqc all.id.txt.summary
# 得到表达矩阵
cat all.id.txt | cut -f1,7- > counts.txt
# 处理表头,/teach/t_rna/要换成自己的路径
less -S all.id.txt |grep -v '#' |cut -f 1,7- |sed 's#/teach/t_rna/project/Human-16-Asthma-Trans/Mapping/Hisat2//##g' |sed 's#.Hisat_aln.sorted.bam##g' >raw_counts.txt
# 列对齐显示
head raw_counts.txt |column -t6.2 salmon
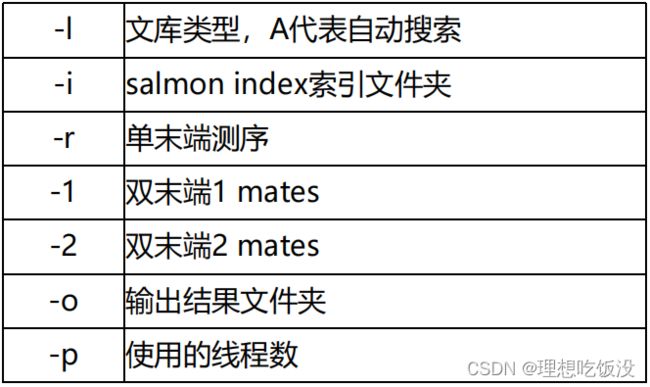
3. 相应的结果如下:
• Name:target transcript 名称,由输入的 transcript database (FASTA file)所提供。
• Length:target transcript 长度,即有多少个核苷酸。
• EffectiveLength:target transcript 计算的有效长度。此项考虑了所有建模的因素,这将影响从这个转录本中取样片段的概率,包括片段长度分布和序列特异性和gc片段偏好
• TPM:估计转录本的表达量
• NumReads:估计比对到每个转录本的reads数
4.步骤如下:
构建索引
cd $HOME/database/genome/GRCh38_release104
# 构建salmon索引
salmon index -t Homo_sapiens.GRCh38.cdna.all.fa.gz -i Homo_sapiens.GRCh38.cdna.all.salmon单个样本
cd $HOME/project/Human-16-Asthma-Trans/Expression/Salmon
# 定义文件夹
index=/home/t_rna/database/genome/GRCh38_release95/Homo_sapiens.GRCh38.cdna.all.salmon/
input=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
outdir=$HOME/project/Human-16-Asthma-Trans/Expression/Salmon
# 单样本测试:\后面不能有空格,否则会报错
salmon quant -i $index -l A -p 5 \
-1 $input/SRR1039510_1_val_1.fq.gz \
-2 $input/SRR1039510_2_val_2.fq.gz \
-o $outdir/SRR1039510.quant多个样本
# 编写脚本,使用salmon批量对目录下所有fastq文件进行定量
# vim salmon.sh
index=/home/t_rna/database/genome/GRCh38_release95/Homo_sapiens.GRCh38.cdna.all.salmon/
input=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
outdir=$HOME/project/Human-16-Asthma-Trans/Expression/Salmon
cat ../../data/cleandata/trim_galore/ID |while read id
do
salmon quant -i ${index} -l A -1 ${input}/${id}_1_val_1.fq.gz -2 ${input}/${id}_2_val_2.fq.gz -p 5 -o ${outdir}/${id}.quant
done
# 后台运行脚本
nohup bash salmon.sh 1>salmon.log 2>&1 &七、差异表达分析
7.1样本异常值与重复性检验
1. 前期准备
rm(list = ls())
## Installing R packages
bioPackages <-c(
"corrplot","ggrepel", # 绘制相关性图形
"stringr", # 处理字符串的包
"FactoMineR","factoextra", # PCA分析软件
"limma","edgeR","DESeq2", # 差异分析的三个软件包
"clusterProfiler", "org.Hs.eg.db", # 安装进行GO和Kegg分析的扩展包
"GSEABase","GSVA" # 安装进行GSEA分析的扩展包
)
## If you are in China, run the command below
options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor/")
options("repos" = c(CRAN="http://mirrors.cloud.tencent.com/CRAN/"))
options(download.file.method = 'libcurl')
options(url.method='libcurl')
# 检查是否设定完毕
getOption("BioC_mirror")
getOption("CRAN")
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
# 安装devtools管理github上的软件包
if (!requireNamespace("devtools", quietly = TRUE)) install.packages("devtools")
## Installing missing packages
lapply( bioPackages,
function( bioPackage ){
if(!bioPackage %in% rownames(installed.packages())){
CRANpackages <- available.packages()
if(bioPackage %in% rownames(CRANpackages)){
install.packages( bioPackage)
}else{
BiocManager::install(bioPackage,suppressUpdates=F,ask=F)
}
}
})
## 验证R包是否安装成功
library(limma)
library(edgeR)
library(DESeq2)
library(FactoMineR)
library(factoextra)
library(clusterProfiler)
library(org.Hs.eg.db)2.过滤去重
rm(list = ls())
options(stringsAsFactors = F)
library(stringr)
## ====================1.读取数据
# 读取raw count表达矩阵
rawcount <- read.table("data/raw_counts.txt",row.names = 1,
sep = "\t", header = T)
colnames(rawcount)
# 查看表达谱
rawcount[1:4,1:4]
# 去除前的基因表达矩阵情况
dim(rawcount)
# 获取分组信息
group <- read.table("data/filereport_read_run_PRJNA229998_tsv.txt",
header = T,sep = "\t",quote = "\"")
colnames(group)
# 提取表达矩阵对应的样本表型信息
group <- group[match(colnames(rawcount),group$run_accession),
c("run_accession","sample_title")]
group
# 差异分析方案为:Dex vs untreated
group_list <- str_split_fixed(group$sample_title,pattern = "_", n=2)[,2]
group_list
## =================== 2.表达矩阵预处理
# 过滤低表达基因
# 1.过滤在至少在75%的样本中都不表达的基因
# 2.过滤平均值count<10的基因
# 3.过滤平均cpm <10 的基因
keep <- rowSums(rawcount>0) >= floor(0.75*ncol(rawcount))
table(keep)
filter_count <- rawcount[keep,]
filter_count[1:4,1:4]
dim(filter_count)
# 加载edgeR包计算counts per millio(cpm) 表达矩阵,并对结果取log2值
library(edgeR)
express_cpm <- log2(cpm(filter_count)+ 1)
express_cpm[1:6,1:6]
# 保存表达矩阵和分组结果
save(filter_count,express_cpm,group_list,
file = "data/Step01-airwayData.Rdata")3.箱线图、小提琴图和概率分布图的绘制
rm(list = ls())
options(stringsAsFactors = F)
# 加载原始表达的数据
lname <- load(file = "data/Step01-airwayData.Rdata")
lname
exprSet <- express_cpm
exprSet[1:6,1:6]
# 样本表达总体分布-箱式图
library(ggplot2)
# 构造绘图数据
data <- data.frame(expression=c(exprSet),
sample=rep(colnames(exprSet),each=nrow(exprSet)))
head(data)
p <- ggplot(data = data,aes(x=sample,y=expression,fill=sample))
p1 <- p + geom_boxplot() + theme(axis.text.x = element_text(angle = 90)) +
xlab(NULL) + ylab("log2(CPM+1)")
p1
# 保存图片
pdf(file = "result/sample_boxplot.pdf",width = 6,height = 8)
print(p1)
dev.off()
# 样本表达总体分布-小提琴图
p2 <- p + geom_violin() +
theme(axis.text = element_text(size = 12),
axis.text.x = element_text(angle = 90)) +
xlab(NULL) + ylab("log2(CPM+1)")
p2
# 保存图片
pdf(file = "result/sample_violin.pdf",width = 6,height = 8)
print(p2)
dev.off()
# 样本表达总体分布-概率密度分布图
m <- ggplot(data=data, aes(x=expression))
p3 <- m + geom_density(aes(fill=sample, colour=sample),alpha = 0.1) +
xlab("log2(CPM+1)")
p3
# 保存图片
pdf(file = "result/sample_density.pdf",width = 7,height = 8)
print(p3)
dev.off()4.主成分分析和热图
# 一键清空
rm(list = ls())
options(stringsAsFactors = F)
# 加载数据并检查
lname <- load(file = 'data/Step01-airwayData.Rdata')
lname
dat <- express_cpm
dat[1:4,1:4]
dim(dat)
## 1.样本之间的相关性-层次聚类树----
sampleTree <- hclust(dist(t(dat)), method = "average")
temp <- as.data.frame(cutree(sampleTree,k = 2))
plot(sampleTree)
pdf(file = "result/sample_Treeplot.pdf",width = 6,height = 8)
plot(sampleTree)
dev.off()
## 2.样本之间的相关性-PCA----
# 第一步,数据预处理
dat <- as.data.frame(t(dat))
dat$group_list <- group_list
# 第二步,绘制PCA图
library(FactoMineR)
library(factoextra)
# 画图仅需要数值型数据,去掉最后一列的分组信息
dat_pca <- PCA(dat[,-ncol(dat)], graph = FALSE)
class(dat_pca)
p <- fviz_pca_ind(dat_pca,
geom.ind = "text", # 只显示点,不显示文字
col.ind = dat$group_list, # 用不同颜色表示分组
palette = c("#00AFBB", "#E7B800"),
addEllipses = T, # 是否圈起来
legend.title = "Groups") + theme_bw()
p
## 3.样本之间的相关性-cor----
exprSet <- express_cpm
library(corrplot)
dim(exprSet)
# 计算相关性
M <- cor(exprSet)
M
g <- corrplot(M,order = "AOE",addCoef.col = "white")
corrplot(M,order = "AOE",type="upper",tl.pos = "d",method = "pie")
corrplot(M,add=TRUE, type="lower", method="number",order="AOE",diag=FALSE,
tl.pos="n", cl.pos="n")
# 绘制样本相关性的热图
library(pheatmap)
anno <- data.frame(sampleType=group_list)
rownames(anno) <- colnames(exprSet)
anno
p <- pheatmap::pheatmap(M,display_numbers = T,
annotation_col = anno,
fontsize = 12,cellheight = 30,
cellwidth = 30,cluster_rows = T,
cluster_cols = T)
p
pdf(file = "result/sample_cor.pdf")
print(p)
dev.off()7.2 差异表达分析方法
7.2.1 limma
分析步骤:
1.创建设计矩阵和对比
2.构建edgeR的DGEList对象,并归一化,拟合模型
3.提取过滤差异分析结果
# 清空当前对象
rm(list = ls())
options(stringsAsFactors = F)
# 读取基因表达矩阵
lname <- load(file = "data/Step01-airwayData.Rdata")
lname
exprSet <- filter_count
# 检查表达谱
dim(exprSet)
exprSet[1:6,1:6]
table(group_list)
# 加载包
library(limma)
library(edgeR)
## 第一步,创建设计矩阵和对比:假设数据符合分布,构建线性模型
# 0代表x线性模型的截距为0
design <- model.matrix(~0+factor(group_list))
colnames(design) <- levels(factor(group_list))
rownames(design) <- colnames(exprSet)
design
# 设置需要进行对比的分组,需要修改
comp <- 'Dex-untreated'
cont.matrix <- makeContrasts(contrasts=c(comp),levels = design)
## 第二步,进行差异表达分析
# 将表达矩阵转换为edgeR的DGEList对象
dge <- DGEList(counts=exprSet)
dge
v <- voom(dge,design,plot=TRUE, normalize="quantile")
fit <- lmFit(v, design)
fit2 <- contrasts.fit(fit,cont.matrix)
fit2 <- eBayes(fit2)
fit2
## 第三步,提取过滤差异分析结果
tmp <- topTable(fit2, coef=comp, n=Inf,adjust.method="BH")
DEG_limma_voom <- na.omit(tmp)
head(DEG_limma_voom)
# 筛选上下调,设定阈值
fc_cutoff <- 2
pvalue <- 0.05
DEG_limma_voom$regulated <- "normal"
loc_up <- intersect(which(DEG_limma_voom$logFC > log2(fc_cutoff) ),
which(DEG_limma_voom$P.Value < pvalue) )
loc_down <- intersect(which(DEG_limma_voom$logFC< (-log2(fc_cutoff))),
which(DEG_limma_voom$P.Value7.2.2 edgeR
分析步骤:
1.创建设计矩阵和对比
2.构建edgeR的DGEList对象,并归一化,拟合模型
3.提取分析结果并筛选显著差异基因
rm(list = ls())
options(stringsAsFactors = F)
# 读取基因表达矩阵信息并查看分组信息和表达矩阵数据
lname <- load(file = "data/Step01-airwayData.Rdata")
lname
exprSet <- filter_count
dim(exprSet)
exprSet[1:4,1:4]
table(group_list)
# 加载包
library(edgeR)
# 构建线性模型。0代表x线性模型的截距为0
design <- model.matrix(~0+factor(group_list))
rownames(design) <- colnames(exprSet)
colnames(design) <- levels(factor(group_list))
design
# 构建edgeR的DGEList对象
DEG <- DGEList(counts=exprSet,
group=factor(group_list))
DEG$samples$lib.size
# 归一化基因表达分布
DEG <- calcNormFactors(DEG)
DEG$samples$norm.factors
# 计算线性模型的参数
DEG <- estimateGLMCommonDisp(DEG,design)
DEG <- estimateGLMTrendedDisp(DEG, design)
DEG <- estimateGLMTagwiseDisp(DEG, design)
# 拟合线性模型
fit <- glmFit(DEG, design)
# 进行差异分析,1,-1意味着前比后
lrt <- glmLRT(fit, contrast=c(1,-1))
# 提取过滤差异分析结果
DEG_edgeR <- as.data.frame(topTags(lrt, n=nrow(DEG)))
head(DEG_edgeR)
# 筛选上下调,设定阈值
fc_cutoff <- 1.5
pvalue <- 0.05
DEG_edgeR$regulated <- "normal"
loc_up <- intersect(which(DEG_edgeR$logFC>log2(fc_cutoff)),
which(DEG_edgeR$PValue7.2.3 DESeq2
分析步骤:
1.构建DESeq2的DESeq对象
2.进行差异表达分析
3.提取差异分析结果,trt组对untrt组的差异分析结果
rm(list = ls())
options(stringsAsFactors = F)
# 读取基因表达矩阵信息
lname <- load(file = "data/Step01-airwayData.Rdata")
lname
# 查看分组信息和表达矩阵数据
exprSet <- filter_count
dim(exprSet)
exprSet[1:6,1:6]
table(group_list)
# 加载包
library(DESeq2)
# 第一步,构建DESeq2的DESeq对象
colData <- data.frame(row.names=colnames(exprSet),
group_list=group_list)
colData
dds <- DESeqDataSetFromMatrix(countData = exprSet,
colData = colData,
design = ~ group_list)
# 第二步,进行差异表达分析
dds2 <- DESeq(dds)
# 提取差异分析结果,trt组对untrt组的差异分析结果
tmp <- results(dds2,contrast=c("group_list","Dex","untreated"))
DEG_DESeq2 <- as.data.frame(tmp[order(tmp$padj),])
head(DEG_DESeq2)
# 去除差异分析结果中包含NA值的行
DEG_DESeq2 = na.omit(DEG_DESeq2)
# 筛选上下调,设定阈值
fc_cutoff <- 1.5
pvalue <- 0.05
DEG_DESeq2$regulated <- "normal"
loc_up <- intersect(which(DEG_DESeq2$log2FoldChange>log2(fc_cutoff)),
which(DEG_DESeq2$padj7.2.4 三种方法的比较
limma,edgeR,DESeq2三大包基本是做转录组差异分析的金标准,大多数转录组的文章都是用这三个R包进行差异分析。
edgeR差异分析速度快,得到的基因数目比较多,假阳性高(实际不差异结果差异)。
DESeq2差异分析速度慢,得到的基因数目比较少,假阴性高(实际差异结果不差异)。
rm(list = ls())
options(stringsAsFactors = F)
# 读取3个软件的差异分析结果
load(file = "data/Step03-limma_voom_nrDEG.Rdata")
load(file = "data/Step03-DESeq2_nrDEG.Rdata")
load(file = "data/Step03-edgeR_nrDEG.Rdata")
ls()
# 提取所有差异表达的基因名
limma_sigGene <- DEG_limma_voom[DEG_limma_voom$regulated!="normal",1]
edgeR_sigGene <- DEG_edgeR[DEG_edgeR$regulated!="normal",1]
DESeq2_sigGene <- DEG_DESeq2[DEG_DESeq2$regulated!="normal",1]
data <- list(limma=limma_sigGene,
edgeR=edgeR_sigGene,
DESeq2=DESeq2_sigGene)
library(VennDiagram)
# 设置颜色
col <- c('#0099CC','#FF6666','#FFCC99')
venn.diagram(data, lwd=1, lty=1, col=col, fill=col,
cat.col=col, cat.cex = 1.8, rotation.degree = 0,
cex=1.5, alpha = 0.5, reverse=TRUE,
width=4000,height = 4000,resolution =600,margin=0.2,
filename="result/DEG_venn.png",imagetype="png")
火山图
rm(list = ls())
options(stringsAsFactors = F)
# 加载原始表达矩阵
load(file = "data/Step01-airwayData.Rdata")
# 读取3个软件的差异分析结果
load(file = "data/Step03-limma_voom_nrDEG.Rdata")
load(file = "data/Step03-DESeq2_nrDEG.Rdata")
load(file = "data//Step03-edgeR_nrDEG.Rdata")
ls()
# 根据需要修改DEG的值
data <- DEG_limma_voom
colnames(data)
# 绘制火山图
library(ggplot2)
colnames(data)
p <- ggplot(data=data, aes(x=logFC, y=-log10(P.Value),color=regulated)) +
geom_point(alpha=0.5, size=1.8) +
theme_set(theme_set(theme_bw(base_size=20))) +
xlab("log2FC") + ylab("-log10(Pvalue)") +
scale_colour_manual(values = c('blue','black','red'))
p
热图
rm(list = ls())
options(stringsAsFactors = F)
# 加载原始表达矩阵
load(file = "data/Step01-airwayData.Rdata")
# 读取3个软件的差异分析结果
load(file = "data/Step03-limma_voom_nrDEG.Rdata")
load(file = "data/Step03-DESeq2_nrDEG.Rdata")
load(file = "data/Step03-edgeR_nrDEG.Rdata")
ls()
# 提取所有差异表达的基因名
limma_sigGene <- DEG_limma_voom[DEG_limma_voom$regulated!="normal",1]
edgeR_sigGene <- DEG_edgeR[DEG_edgeR$regulated!="normal",1]
DESeq2_sigGene <- DEG_DESeq2[DEG_DESeq2$regulated!="normal",1]
head(DESeq2_sigGene)
# 绘制热图
dat <- express_cpm[match(limma_sigGene,rownames(express_cpm)),]
dat[1:4,1:4]
group <- data.frame(group=group_list)
rownames(group)=colnames(dat)
group
# 加载包
library(pheatmap)
p <- pheatmap(dat,scale = "row",show_colnames =T,show_rownames = F,
cluster_cols = F,
annotation_col=group,
main = "limma's DEG")
group
dex_exp <- express_cpm[,match(rownames(group)[which(group$group=="Dex")],
colnames(express_cpm))]
untreated_exp <- express_cpm[,match(rownames(group)[which(group$group=="untreated")],
colnames(express_cpm))]
data_new <- cbind(dex_exp, untreated_exp)
dat1 <- data_new[match(limma_sigGene,rownames(data_new)),]
p <- pheatmap(dat1, scale = "row",show_colnames =T,show_rownames = F,
cluster_cols = F,
annotation_col=group,
main = "limma's DEG")7.3 功能注释和功能富集
7.3.1 功能注释
rm(list = ls())
options(stringsAsFactors = F)
library(clusterProfiler)
library(org.Hs.eg.db)
# 读取3个软件的差异分析结果
load("data/Step01-airwayData.Rdata")
load(file = "data/Step03-limma_voom_nrDEG.Rdata")
load(file = "data/Step03-DESeq2_nrDEG.Rdata")
load(file = "data/Step03-edgeR_nrDEG.Rdata")
ls()
# 提取所有差异表达的基因名
limma_sigGene <- DEG_limma_voom[DEG_limma_voom$regulated!="normal",1]
edgeR_sigGene <- DEG_edgeR[DEG_edgeR$regulated!="normal",1]
DESeq2_sigGene <- DEG_DESeq2[DEG_DESeq2$regulated!="normal",1]
# 根据需要更改DEG的值
DEG <- limma_sigGene
head(DEG)
gene_all <- rownames(filter_count)
#### 第一步,从org.Hs.eg.db提取ENSG的ID 和GI号对应关系
keytypes(org.Hs.eg.db)
# bitr in clusterProfiler
allID <- bitr(gene_all, fromType = "ENSEMBL",
toType = c( "ENTREZID" ),
OrgDb = org.Hs.eg.db )
head(allID)
degID <- bitr(DEG, fromType = "ENSEMBL",
toType = c( "ENTREZID" ),
OrgDb = org.Hs.eg.db )
head(degID)
# KEGG analysis----
# 设置pvalue与qvalue为最大值,输出所有结果,
# 然后根据结果来筛选显著性通路,
# 这样就不必因为没有显著性结果重新跑一边富集过程
enrich <- enrichKEGG(gene = degID[,2],
organism='hsa',
universe=allID[,2],
pvalueCutoff=1,
qvalueCutoff=1)
# 计算富集因子
GeneRatio <- as.numeric(lapply(strsplit(enrich$GeneRatio,split="/"),function(x)
as.numeric(x[1])/as.numeric(x[2])))
head(GeneRatio)
BgRatio <- as.numeric(lapply(strsplit(enrich$BgRatio,split="/"),function(x)
as.numeric(x[1])/as.numeric(x[2]) ))
head(BgRatio)
enrich_factor <- GeneRatio/BgRatio
out <- data.frame(enrich$ID,
enrich$Description,
enrich$GeneRatio,
enrich$BgRatio,
round(enrich_factor,2),
enrich$pvalue,
enrich$qvalue,
enrich$geneID)
colnames(out) <- c("ID","Description","GeneRatio","BgRatio","enrich_factor","pvalue","qvalue","geneID")
write.table(out,"result/trut_VS_untrt_enrich_KEGG.xls",row.names = F,sep="\t",quote = F)
out_sig0.05 <- out[out$qvalue<0.01,]
# barplot
bar <- barplot(enrich,showCategory=20,title="KEGG Pathway",
colorBy="p.adjust")
bar
# 保存
pdf(file = "result/kegg_bar_plot.pdf",width = 8,height = 6)
print(bar)
dev.off()
# dotplot
dot <- dotplot(enrich,x="geneRatio",showCategory=10,font.size=12,title="KEGG Pathway")
dot
# 保存
pdf(file = "result/kegg_dot_plot.pdf",width = 8,height = 6)
print(dot)
dev.off()
# GO
enrich <- enrichGO(gene =degID[,2],OrgDb='org.Hs.eg.db',
ont="BP",universe=allID[,2],pvalueCutoff=1,qvalueCutoff=1)
# 计算富集因子
GeneRatio <- as.numeric(lapply(strsplit(enrich$GeneRatio,split="/"),function(x)
as.numeric(x[1])/as.numeric(x[2])))
BgRatio <- as.numeric(lapply(strsplit(enrich$BgRatio,split="/"),function(x)
as.numeric(x[1])/as.numeric(x[2])))
enrich_factor <- GeneRatio/BgRatio
out <- data.frame(enrich$ID,
enrich$Description,
enrich$GeneRatio,
enrich$BgRatio,
round(enrich_factor,2),
enrich$pvalue,
enrich$qvalue,
enrich$geneID)
colnames(out) <- c("ID","Description","GeneRatio","BgRatio","enrich_factor","pvalue","qvalue","geneID")
write.table(out,"result/trut_VS_untrt_enrich_GO.xls",row.names = F,sep="\t",quote = F)
out_sig0.05 <- out[out$qvalue<0.01,]
# barplot
bar <- barplot(enrich,showCategory=10,title="Biological Pathway",colorBy="p.adjust")
bar
# 保存
pdf(file = "result/BP_bar_plot.pdf",width = 6,height = 6)
print(bar)
dev.off()
# dotplot
dot <- dotplot(enrich,x="geneRatio",showCategory=10,font.size=12,title="Biological Pathway")
dot
# 保存
pdf(file = "result/BP_dot_plot.pdf",width = 6,height = 6)
print(dot)
dev.off()7.3.2 功能富集GSEA
# 清空当前环境变量
rm(list = ls())
options(stringsAsFactors = F)
# 加载包
library(GSEABase)
library(clusterProfiler)
# 加载数据
lnames <- load("data/Step03-limma_voom_nrDEG.Rdata")
lnames
DEG <- DEG_limma_voom
## 构造GSEA分析数据
# 去掉没有配对上symbol的行
DEG <- DEG[DEG$SYMBOL!="NA",]
geneList <- DEG$logFC
names(geneList) <- DEG$SYMBOL
head(geneList)
geneList <- sort(geneList,decreasing = T)
head(geneList)
# 选择gmt文件(MigDB中的全部基因集)
geneset <- read.gmt("data/MsigDB/v7.1/c2.cp.kegg.v7.1.symbols.gmt")
egmt <- GSEA(geneList,
TERM2GENE=geneset,
verbose=T,pvalueCutoff = 1)
kegg_gsea <- as.data.frame(egmt@result)
colnames(kegg_gsea)
write.table(kegg_gsea,"result/gsea_kegg_fc.xls",
row.names = F,sep="\t",quote = F)
library(enrichplot)
gseaplot2(egmt, "KEGG_RIBOSOME",
title = "KEGG_RIBOSOME",
pvalue_table = T,color = "red")7.3.3 功能富集GSVA
rm(list = ls())
options(stringsAsFactors = F)
## 读取基因表达矩阵
lnames <- load(file = "data/Step01-airwayData.Rdata")
lnames
## 将表达矩阵的ensembl ID换成gene symbol
library(org.Hs.eg.db)
keytypes(org.Hs.eg.db)
library(clusterProfiler)
id2symbol <- bitr(rownames(express_cpm),
fromType = "ENSEMBL",
toType = "SYMBOL", OrgDb = org.Hs.eg.db )
head(id2symbol)
express_cpm <- data.frame(GeneID=rownames(express_cpm),express_cpm)
express_cpm[1:4,1:4]
express_cpm <- merge(id2symbol, express_cpm, by.x="ENSEMBL", by.y="GeneID")
dim(express_cpm)
# 处理多个ID对一个symbol的情况
library(limma)
exprSet <- limma::avereps(express_cpm[,-c(1:2)],ID=express_cpm[,2])
dim(exprSet)
exprSet[1:4,1:4]
## 将表达矩阵转换成通路矩阵
library(GSEABase)
library(GSVA)
geneset <- getGmt("data/MsigDB/v7.1/c2.cp.kegg.v7.1.symbols.gmt")
class(geneset)
es_max <- gsva(exprSet, geneset, mx.diff=F, verbose=T, parallel.sz=8)
dim(es_max)
## 做差异分析
library(limma)
design <- model.matrix(~0+factor(group_list))
colnames(design) <- levels(factor(group_list))
rownames(design) <- colnames(es_max)
design
contrast.matrix <- makeContrasts("Dex-untreated",levels = design)
contrast.matrix
fit <- lmFit(es_max,design)
fit2 <- contrasts.fit(fit, contrast.matrix)
fit2 <- eBayes(fit2)
res <- decideTests(fit2, p.value=0.05)
summary(res)
tempOutput <- topTable(fit2, coef=1, n=Inf)
nrDEG <- na.omit(tempOutput)
head(nrDEG)
# 得到显著通路
nrDEG_sig <- nrDEG[nrDEG$P.Value<0.01,]
## barplot
library(pheatmap)
library(stringr)
data <- es_max[match(rownames(nrDEG_sig),rownames(es_max)),]
rownames(data) <- gsub("KEGG_","",rownames(data))
anno <- data.frame(group=group_list)
rownames(anno) <- colnames(data)
p <- pheatmap::pheatmap(data, fontsize_row = 8,
height = 11,annotation_col = anno,show_colnames = F)