【无标题】Deep AVPpred:人工智能驱动的病毒感染多肽药物的发现
《Deep-AVPpred: Artificial intelligence driven discovery of peptide drugs for viral infections》
单位: 印度瓦拉纳西州BHU印度理工学院计算机科学与工程系
- 作者: Ritesh Sharma, Sameer Shrivastava, Sanjay Kumar Singh, Senior Member, IEEE, Abhinav Kumar,Amit Kumar Singh, Senior Member, IEEE, and Sonal Saxena
- 发表时间:2021 年 11 月 25 日
- 发表期刊: IEEE Journal of Biomedical and Health Informatics

paper :https://ieeexplore.ieee.org/abstract/document/9627580
web:https://deep-avppred.anvil.app/
Abstract
病毒迅速增加暴发导致病毒疾病在不同物种和地理边界之间传播。人畜共患病2019冠状病毒疾病已经极大地影响了人类的福祉,而COVID-19流行病就是一个燃烧的例子。现有的抗病毒药物疗效低,副作用严重,毒性高,市场供应有限。因此,人们对天然物质进行了抗病毒活性测试。抗病毒肽(AVPs)等宿主防御分子存在于动植物中,保护它们免受病毒入侵。然而,从天然来源获取AVP用于制备合成肽药物既昂贵又耗时。因此,需要一个insilico模型来识别新的AVP。我们提出了Deep-AVPpred,这是一种用于发现蛋白质序列中AVP的深度学习分类器,它利用了迁移学习的概念和深度学习算法。该分类器的性能优于现有的分类器,在验证集和测试集上分别达到了约94%和93%的精度。高精度表明,Deep-AVPpred可用于提出新的AVP用于合成和实验。通过利用深度AVPpred,我们在人类干扰素-α家族蛋白中发现了新的AVP。这些AVP可以化学合成,并通过实验验证其对不同病毒的抗病毒活性。DeepAVPpred部署为web服务器,可在https://deep-avppred.anvil.app/,可用于预测新的AVP,用于开发用于人类和兽医的抗病毒化合物。
索引项:抗病毒肽,2019冠状病毒,深度学习,迁移学习,干扰素,人畜共患病病毒
I. INTRODUCTION
在过去40年中,世界上出现了许多病毒暴发,包括1981年的艾滋病毒、2002年的SARS-CoV、2009年的H1N1流感病毒、2012年的MERS-CoV、2013年的埃博拉病毒和2019年的SARS-CoV-2。这种频繁出现和再次出现的病毒暴发严重影响了人类的福祉,并对生命和财产造成了重大的损失。。由于常用抗病毒药物的市场供应有限、严重副作用和高毒性,控制病毒性疾病具有挑战性。
因此,许多天然化合物都经过了抗病毒活性测试。抗病毒肽(AVPs)天然存在于动植物中,在杀死入侵病毒方面发挥着重要作用。这些AVP的抗病毒作用机制包括:(1)抑制病毒附着在宿主细胞受体上。(2) 防止病毒与宿主细胞共受体相互作用。(3) 抑制病毒包膜与宿主细胞膜的融合。(4) 通过与病毒核酸相互作用抑制病毒复制。(5) 参与某些病毒蛋白质的翻译后修饰过程;(6)干扰病毒颗粒组装。大多数AVP通过上述机制(单独或联合)降低病毒载量。与常用的抗病毒药物相比,AVP有很多好处,比如它们是天然的,可以通过多种方式杀死病毒,副作用较小,对宿主细胞的毒性较小。因此,AVPs作为传统化学抗病毒药物的替代品,最近受到了广泛关注。尽管各种生物都会产生AVP,但从自然资源中寻找有效的AVP既耗时又昂贵。因此,有必要开发一种电子工具来发现蛋白质序列中新的AVPs。
诸如AVPpred、IAMPRED、MetaiAVP、AVPIden、ENNAVIA等分类器在文献中可用作网络服务器,可用于从自然来源识别AVP。这些分类器利用了机器学习算法和人工神经网络的各种手工特征。随着时间和技术的进步,大量的AVP现在可以在数据库中使用,使我们能够应用深度学习算法。与机器学习相比,深度学习算法具有以下几个优点:(i)深度学习算法可以自动从数据中提取最佳特征,消除我们对领域专家的依赖。(ii)深度学习提取的特征通常优于手工制作的特征。因此,深度学习算法通常优于机器学习算法。(iii)迁移学习的概念很容易适用于深度学习算法,这使他们能够利用从之前任务中获得的知识来提高绩效。此外,深度学习模型已经在各种应用中证明了它们的能力。
因此,在本文中,我们提出了Deep-AVPpred,这是一种结合迁移学习和深度学习的深度学习算法。该模型采用一维卷积神经网络,利用不同尺度的多个核,迁移学习的概念通过预训练嵌入实现。[20]中的作者通过以无监督的方式对UniRef50中数百万蛋白质序列的33层变换器模型进行训练,获得了这些预训练的嵌入。
我们比较了将提出的模型(Deep-A VPpred)与现有模型(A VPpred、IAMPRED、Meta iA VP、A VPIden、ENNA VIA)的性能进行了比较,发现其性能优于其他模型,在验证集和测试集上分别达到了约94%和93%的精度。Deep-A VPpred 实验表明,高精度的Deep-A VPpred 可以用于深部合成。干扰素在人类和其他脊椎动物中起着宿主防御蛋白的作用,以抵御入侵的病毒。因此,我们利用我们提出的模型,在属于人类干扰素-α家族的12种抗病毒蛋白质中发现了新的VPs。基于特定的选择标准,我们从每个干扰素蛋白质中提出一个AVP,用于湿实验室合成和抗病毒活性评估。此外,该模型被部署为一个网络服务器,可以免费在线获得,以帮助研究人员进行AVP预测。该服务器可用于在蛋白质序列中发现新的AVP,结果以报告的形式呈现,其中包括预测的肽及其物理化学性质。
本文的主要贡献如下:
1)我们提出了Deep-AVPpred,这是一种用于识别蛋白质序列中AVP的深度学习分类器,其性能优于现有的AVP分类器,在验证集和测试集上分别达到约94%和93%的精度。
2) 我们提出的模型可以自动从肽中提取最佳特征,消除了我们对领域专家生成最佳特征的依赖。
3) 我们使用Deep-AVPpred来筛选属于人类干扰素-α家族的抗病毒蛋白质,并鉴定出可以在实验室进行化学合成的新型AVP,并对其抗病毒活性进行评估。
4) 该模型被部署为一个网络服务器,以帮助研究人员从蛋白质序列中发现新的AVP.
本文的其余部分组织如下。第二节提供了有关数据集和拟议框架的信息。第三节给出了从我们提出的模型中获得的结果及其与其他最先进分类器的比较。第四节介绍了利用我们提出的模型在人类干扰素-α家族蛋白质中识别新的AVP,第五节给出了结论。
II. MATERIALS AND METHODS
A. Dataset
在目前的研究中,我们收集了10203个长度为∈ [5,50]来自VPpred、DBAASP、DRAMP、SA TPDB和StarPep。8792个非A VP来自VPpred和瑞士Prot。我们使用类似的方法从Swiss-Prot获得了非A VPs在之前的论文中。对Swiss-Prot进行了查询,以获得经过审查的、人工注释的长度蛋白质∈ [5,50]不包含以下任何关键词:抗病毒、抗真菌、抗菌、抗菌、抗生素、抗毒素、抗肿瘤、防御素、抗结核、抗HIV、抗疟疾、抗癌、抗内毒素、抗糖尿病、杀虫、细胞因子、抗氧化剂、抗RSA、抗RAM阳性、抗RAM阴性、抗质子、抗质子、细菌素、抗生物膜、抗炎、,抗寄生虫,分泌,排泄,效应物。在收集AVP和非AVP后,我们应用了以下预处理步骤:(i)删除重复序列。(ii)删除含有非天然氨基酸的序列。(iii)消除了以AVP和非AVP形式出现的序列。(iv)使用了阈值为0.7的CD-HIT-2D程序,消除了与AVP至少70%相同的非AVP。(v) 确定了至少20个AVP中存在的四个长度基序,并消除了包含这些基序的非AVP。
预处理后,我们得到4432个非AVP和4090个AVP。为了使数据集平衡,我们删除了342个非AVP。因此,最终数据集(Ds)包含8180个肽(AVPs:4090,非AVPs:4090)。所有来自Ds的4090 A VP都有证据表明对至少一种病毒有效。然而,DSA的4090个非A VP中没有一个能有效对抗任何病毒的证据。我们进一步将包含4090个A VP和4090个非A VP的DSS分为三组,即训练集( S T r a i n S^{T rain} STrain)、V验证集(S V a l ^{V al} Val)和测试集(S T e s t l ^{Testl} Testl)。 S T r a i n S^{T rain} STrain含有60%的肽,可定义如下:

我们利用S V a l ^{V al} Val来调整超参数。保留了从调整过程中获得的最佳模型,并将其命名为Deep-AVPpred。为了确定Deep-A VPpred的稳定性,我们又对其进行了四次训练,表1中提供了五次运行验证数据所得分数的平均值±标准偏差值。S T e s t l ^{Testl} Testl既不用于训练,也不用于超参数选择;因此,它被用来以一种无偏见的方式将我们提出的模型和最先进的模型进行比较。

B. Proposed framework
Deep-AVPpred的拟议框架如图1所示。深度学习算法只能处理数值数据。因此,我们在将原始肽序列输入框架之前,将其转换为数值。为此,我们使用预训练的嵌入物,使用长度为1280的载体对肽的每个氨基酸进行编码。
该框架的第一层是输入层,我们通过输入层输入数据(编码肽)。通过该层传送的数据必须具有相同的维度。我们已经考虑了长度的肽∈ [5,50]; 因此,肽长度的最大值为50。因此,我们对长度小于50的编码肽使用长度为1280的零载体进行后填充。

单个卷积核不足以识别分类所需的不同模式。我们需要多个不同大小的卷积核·,以了解氨基酸之间的不同关系。因此,我们将输入层的输出输入到四个1D卷积层。与这些层一起使用的内核大小分别为4、5、6和7。每个内核大小的过滤器数量是200。这些过滤器有效地捕获了4、5、6和7个氨基酸序列组中的模式。每个1D卷积层都涉及一个基本操作,即编码肽和不同过滤器之间的1D卷积操作。卷积运算后,使用校正线性单元(ReLU)激活函数,其定义如下:

从每个1D卷积层中,我们获得了200个特征图。
在每个1D卷积层之后,我们应用1D全局最大池层。该层的工作是通过从特征图中获取最大值来完成下采样。从每个1D全局最大池层,我们得到了200个单变量向量。
接下来,将从四个1D Global Max Pooling层中的每一层获得的单变量向量连接起来,这为我们提供了长度为800的特征向量。
一个独立的层(ICL),然后是一个独立的退出率。使用30。ICL的概念最早是在[31]中引入的,作者结合了两种流行的技术,批量标准化和退出(批量标准化后退出),来构建ICL。他们进行了大量测试,发现在重量层之前使用ICL可以带来更稳定的训练、更快的收敛速度和更好的泛化性能。
ICL后,使用三个致密层,分别由64、32和8个神经元组成,具有ReLU激活功能。
最后,应用由单个神经元组成的密集层,该神经元具有一个S形激活函数(见等式5),输出一个值∈ [0,1]. 如果价值∈ [0,0.5],肽属于非AVP类;否则,它属于AVP类。
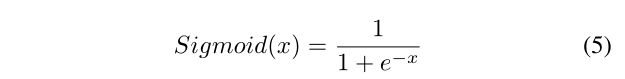
使用自适应矩估计(Adam)优化器更新网络权重。在Adam中,分别采用了梯度GT的指数移动平均值MTA和VT以及梯度g2的平方T。MTA和vt都被初始化为零向量,MTA和vt的值在每次迭代t中都得到更新。每次迭代中考虑的训练样本数为8(即批量=8)。Adam对每次迭代t的参数更新如下:
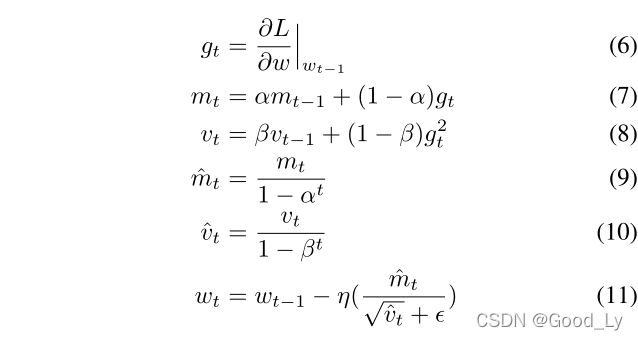
其中L是二元交叉熵损失函数?是一个用于防止被零除的小数字,α和β分别控制mt和vt的衰减率(两者的默认值接近1),η表示学习率,η的最佳值为10−5.
II. RESULTS
所有实验都是在一个CPU计算节点上进行的,该节点具有2。4 GHz英特尔至强天湖6148处理器和192 GB内存。使用深度学习lib库实现了深度学习算法[32 ]。使用众所周知的分类评估指标评估模型的性能,即准确度(Acc)、敏感性(Sn)、精确度(Pr)、F1评分(Fs)、特异性(Sp)、ROC曲线下面积(AUROC)。
1) Results obtained from our proposed model and other state-of-the-art models
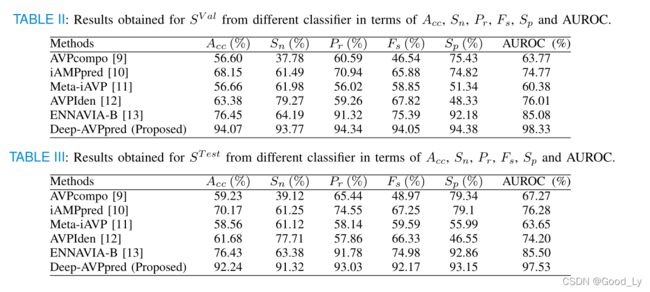
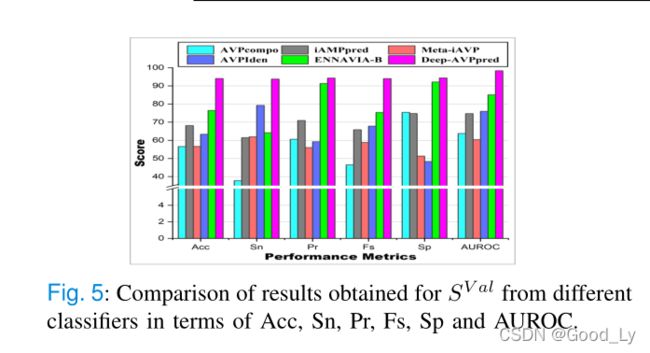
表二、图2和表三、图3分别显示了我们提出的模型和其他现有模型在 S V a l S^{V al} SVal和 S S S{T est}$上的结果。
对于AVPpred和ENNAVIA,服务器中提供了多种机器学习模型。因此,我们为他们提供了可用模型中性能最佳的模型的结果。ENNAVIA server不提供长度小于7且大于40的肽的预测,因此我们考虑了具有长度的序列∈[7,40]同时从中进行预测。类似地,VPIden服务器不提供长度小于8的肽的预测,因此我们考虑了具有长度的序列∈[8,50]同时从中进行预测。


2) Comparison using Validation set ( S V a l S^{Val} SVal)
从图4和图5可以看出,我们提出的模型Deep-A VPpred在SV al上的性能优于其他模型。在Acc、Pr、Fs、Sp和AUROC方面,表现第二好的是Ennavia-B,它实现的Acc、Pr、Fs、Sp和AUROC值比我们提出的模型低近18%、3%、19%、2%和13%。在Sn方面表现第二好的是VPIden,它的Snvalue比我们提出的模型低近15%。

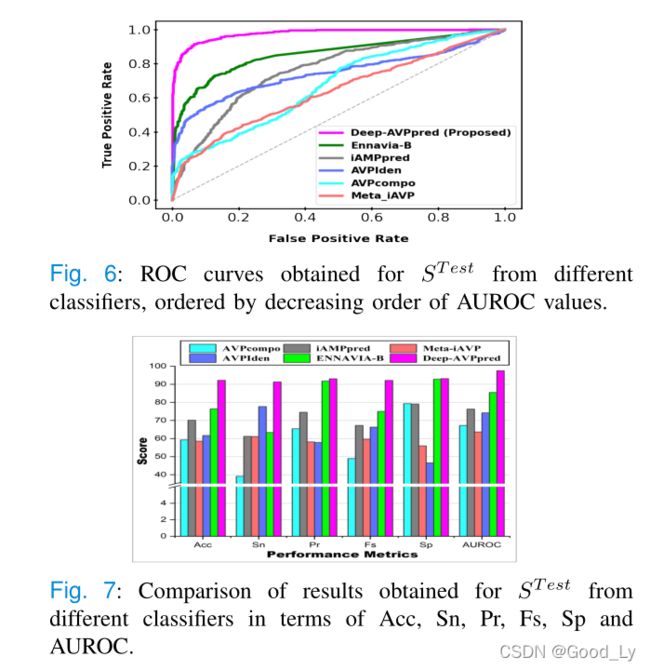
3) Comparison using Test set ( STest)
ST既不用于训练也不用于超参数选择;因此,我们利用它以无偏见的方式将我们提出的模型与最先进的模型进行比较。如图6和图7所示,我们提出的模型Deep-A VPpred也比其他模型表现得更好。在Acc、Pr、FSA和AUROC方面排名第二的是Ennavia-B,它实现了Acc、Pr、Fs、 AUROC值比我们提出的模型低近16%、1%、17%和12%(Ennavia-B和DeepA VPpred在SPI方面的性能大致相当)。在Sn方面表现第二好的是VPIden,它的Snvalue比我们提出的模型DeepA VPpred低近14%。
4) Density Plots
为了显示模型的置信度和预测时的错误分类,我们提供了密度图(使用seaborn library[33]的kdeplot函数绘制)。每个模型提供两个密度图(一个对应于AVP预测,另一个对应于非AVP预测)。我们考虑了SV aland ST EST中可用肽的模型预测,以生成密度图。
以下标准用于使用密度图估计性能:1)重叠标准:减小与VPs对应的密度图之间的重叠,以及非A VPs的错误分类越少。从图8可以看出,我们提出的模型Deep-A VPpred在对应于A VPs和非A VPs的密度图之间的重叠最小,这意味着Deep-A VPpred的错误分类最少。2) 峰值标准:对其预测有信心的模型正好有两个峰值,一个接近0(非AVP),一个接近1(AVP)。只有我们提出的模型Deep AVPpred满足峰值标准,如图8所示,分别对应于nonA VPs和A VPs的密度图显示了两个峰值,一个接近0,另一个接近1。除了我们提出的模型外,对于非a VPs,ENNA VIA-B通过显示接近零的峰值部分满足峰值标准。

IV. PREDICTION OF AVPS IN THE ANTIVIRAL PROTEINS
使用Deep-A VPpred,我们在人干扰素(IFN)中发现了新的AVP(见表IV)。干扰素是一种具有强大抗病毒活性的糖蛋白,是抵御入侵病原体的最初防线之一。根据其遗传、结构和功能特性,以及细胞表面的受体,干扰素分为三种类型:I型、II型和III型。其中,I型干扰素是最大的一类,包括IFN-α、IFN-β、IFN-β、?,IFN-ω、IFN-κ、IFN-δ、IFN-τ和IFN-ζ。从I型干扰素中,我们选择了人类干扰素-α家族来鉴定新的VPs。它包含13个基因,编码12种蛋白质,其中IFN-α-13与IFN-α-1相同。人类IFN-α家族蛋白因其对不同病毒的抗病毒活性而闻名,包括人类偏肺病毒、水泡性口炎病毒、猪瘟病毒、甲型流感病毒、乙型肝炎病毒、丙型肝炎病毒、戊型肝炎病毒和人类免疫缺陷病毒-1。
我们从NCBI[40]的蛋白质数据库中获得了人类IFN-α家族蛋白质,并执行以下步骤来识别VPs:
1) 肽库的建立:通过获得长度的子串设计肽库∈ [10,20]来自每个蛋白质序列。
2) 基于概率阈值的肽选择:用肽库中的肽喂养Deep-AVPpred。只有DeepA VPpred提供的肽≥ 考虑0.99。
3) 模体搜索:我们发现445个长度为4的模体,只存在于20个或更多的A VP序列中。这445个基序跨越了4090个VP中的1908个,约占VP的47%。我们提取了456个不同的3长度图案长度为3的子串来自445,4个长度的图案。这3个长度基序包含1461个额外的A VP序列,这4个长度基序未涵盖。因此,3和4个长度基序跨越3369个VP,约占VP的82%。
我们检查了从上一步获得的肽中是否存在这3个和4个长度基序。根据检测的4090A VPs中长度分别为3和4的基序的出现情况,计算每个肽的得分3和得分4。例如,如果肽P包含两个基序M1和M2,则长度均为3,得分3可通过将M1和M2在所考虑的4090 A VP中的出现次数相加而获得,类似地,得分4可获得。我们忽略了3分和4分都为零的序列。
4) 溶解度:药物分子要分布在全身并达到其目标,必须溶于体液。因此,我们使用PepCalc测试了从上一步获得的序列的水溶性(https://pepcalc.com/),忽略了水溶性差的序列。
5) 分类:最后,我们按照得分4和得分3值递减的顺序(如果得分4相同,则根据得分3进行排序)对上一步获得的肽进行排序,并从每个蛋白质中提出独特的肽,该肽位于湿实验室合成和实验的最顶端,如表IV所示。肽(qslekfstelyqlndlea)是在多种蛋白质(IFN-α-4/7/10)中预测的最顶端的肽,在表的最后一行中单独列出。
为了支持湿实验室的研究人员从任何蛋白质中识别出新的VPs,我们在https://deep-avppred.铁砧app/。我们已经使这个web服务器变得灵活,用户可以根据需要定制各种参数。这些参数包括:
1)长度:制备肽库时考虑的长度。这可能是长度∈ [5,50](默认值:长度=20)。
2) 概率:概率的阈值。概率大于0的任何值。5(默认值:0.99)。
3) Motif:是/否,表示是否进行Motif搜索以获得分数3和分数4的值。(默认值:是)。
4)溶解度:是/否,表示是否考虑水溶解度(默认值:是)。
一旦用户输入蛋白质序列和定制参数,网络服务器将生成报告,以下信息:(i)肽序列(ii)肽的长度(iii)肽的分子量(iv)肽的净电荷(v)水溶性(好/差)(vi)概率(vii)基序3:肽中存在长度为3的不同基序(viii)基序4:肽中存在长度为4的不同基序(ix)得分3(x)得分4。
V. CONCLUSION
不同病毒的暴发除了扰乱自然之外,还造成人类和动物生命的重大损失,影响人们的福祉。病毒感染使用不同的抗病毒药物进行治疗,但大多数药物的市场供应有限,副作用严重,毒性高,这使得病毒感染的管理成为一项具有挑战性的任务。一些来自天然来源、合成和半合成的化合物因其抗病毒特性而被探索。AVP是一类分子,具有抗病毒特性,副作用小,毒性小,并以多种方式杀死病毒。因此,人们对探索和发现新的AVP作为传统抗病毒药物的替代品越来越感兴趣。生物体产生AVP作为一种自然防御,但从自然来源寻找有效的AVP既耗时又耗费大量成本。在文献中,像AVPpred、IAMPRED、MetaiAVP、AVPIden、ENNAVIA这样的分类器可以作为网络服务器,用于预测AVP。这些分类器利用了机器学习算法和人工神经网络的各种手工特征。随着时间和技术的进步,数据库中现在有足够的AVP。因此,可以使用深度学习算法,与机器学习算法相比,深度学习算法有几个优点,包括更好的性能和不依赖领域专家构建最佳特征。因此,在当前的工作中,我们提出了一个名为Deep-AVPpred的深度学习模型。为了开发该模型,我们采用了迁移学习和深度学习算法。我们将我们提出的模型与其他模型的性能进行了比较,发现它优于其他模型,在 S V a l 和 S T e s t S^{Val}和 S^{Test} SVal和STest上分别实现了约94%和93%的精度。我们还从属于人类干扰素-α家族的抗病毒蛋白质中鉴定出了新的AVP,并根据特定的选择标准建议从这些蛋白质中提取VPs用于湿实验室合成和评估。建议的模型也被部署为web服务器,可在https://deep-avppred.铁砧app/。该服务器可用于识别蛋白质序列中的新VPs,研究结果以报告的形式呈现,其中包括预测的肽及其物理化学性质。
该模型基于AVP序列进行训练。培训期间不提供特定于病毒的信息。因此,该模型将以AVP/非AVP的形式提供输出(即,预测的肽将作为AVP或不作为AVP),而不考虑其可针对的病毒。将来,当在不同病毒上测试的AVP有足够的数据可用时;这项工作可以扩展到两个阶段建立级联模型。第一阶段可以以AVP/非AVP的形式提供输出,而第二阶段可以提供关于第一阶段中识别的AVP可能针对的病毒的见解。在目前的工作中,我们考虑了人类IFN-α家族来识别VPs。类似地,将来也可以考虑使用其他干扰素来确定新的VPs。虽然我们在当前的研究中考虑了不同数据库中可用的所有VP,但有通过加入未来可能提供的附加AVPs,仍有改进拟议模型性能的空间。
ACKNOWLEDGEMENT
感谢IVRI的NASF和IIT(BHU)的PARAM Shivay超级计算设施提供的支持和资源。
网站工具:https://anvil.works/login