蛋白质折叠
文章目录
- 4. GNNs for Protein folding
-
- Chemical Structures as Graphs
- Protein Structure Prediction
-
- Methods for Protein Structure Prediction
- Old method: fragment assembly
- New Strategy
-
- Co-evolution Analysis
- Towards An End-to-End Workflow
- AlphaFold2 architecture
-
- 补充:跟李沐精读论文
- 补充:钟博子韬同学的报告
-
- Multimer模型对输入进行改进
- 预测结果
4. GNNs for Protein folding
Chemical Structures as Graphs

- 化学结构作为图: 化学分子的结构可以看作是图,其中原子被看作是顶点,化学键被看作是边。这种视角为化学分子的计算和分析提供了强大的工具,特别是在计算化学和药物设计等领域。
- 分子的结构相似性: 分子的结构相似性是通过比较两个或更多分子的化学结构来度量的。这种相似性通常是通过比较分子图的特性,如原子类型,化学键类型,原子间的连接关系等来确定的。
- 图论与化学: 图论是数学的一个分支,主要研究图的属性和结构。在化学中,图论被广泛应用于分子结构的描述和分析。通过将分子视为图,可以使用图论的概念和技术来解决许多化学问题,比如判断两个分子是否同构,预测分子的化学反应性等。
- Weisfeiler-Lehman测试: Weisfeiler-Lehman (WL) 测试是一种用于确定两个图是否同构的算法。同构的图在结构上是相同的,即使顶点的标签或排列顺序不同。WL测试通过一种迭代的过程来对图的顶点进行重新标记,如果两个图在经过足够多次迭代后得到的标记相同,那么这两个图就被认为是同构的。在化学中,WL测试可以被用来比较分子的结构,从而帮助化学家识别结构相似的分子。

简单解释一下,我们看第一排:
- 第一张图蓝色的,所有节点一样
- 我们把有两个邻居的节点标为绿色,有三个邻居的节点标为黄色,我们就得到了第二张图
- 然后,我们把有两个黄色邻居的节点标为紫色,把有二绿一黄邻居的节点标为橙色,把有一绿一黄邻居的节点标为灰色
- 最后我们统计这个分子图的节点就是一紫二灰二橙。
- 我们发现下面这个分子图虽然长的跟上面这个不一样,但是通过这样简单计算出来也是一紫二灰二橙。我们就认为是同构的。

这段内容概述了利用图神经网络进行药物研发的两个主要技术:虚拟筛选/分子属性预测和新药设计。
虚拟筛选/分子属性预测: 虚拟筛选是一种计算技术,用于在大规模化合物库中识别可能的药物候选物。在这个过程中,图神经网络被用于学习分子的低维度表示,这些表示可以用于预测分子的属性,如溶解度、毒性等。这项技术已经在多项研究中得到应用,包括Duvenaud等人(2015),Kearnes等人(2016)和Jin等人(2018)的研究。
新药设计: 新药设计,也被称为"de novo"药物设计,是一个利用计算工具来设计新的潜在药物的过程。在这个过程中,图神经网络被用于生成新的分子结构。这项技术已经在多项研究中得到应用,包括Olivecrona等人(2018),Gomez-bombarelli等人(2018),Jin等人(2018)和Popova等人(2018)的研究
Protein Structure Prediction
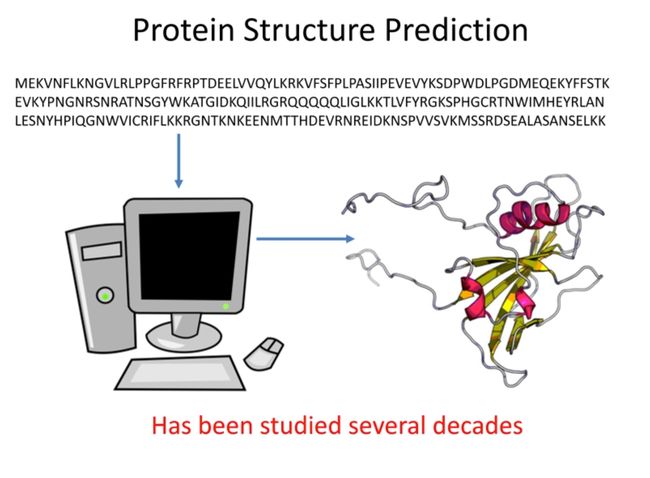
三维的蛋白质结构由一维的氨基酸序列折叠而成,这个问题已经被研究了几十年。
蛋白质结构预测是一种用于确定蛋白质的三维结构的方法,通常是通过其氨基酸序列。理解蛋白质的三维结构是非常重要的,因为它可以帮助我们理解蛋白质的功能,并在药物设计中找到可能的药物靶点。
蛋白质折叠通常涉及如下几个主要的结构元素:
- α-螺旋(Alpha-helices): α-螺旋是蛋白质的一种常见二级结构,由于胺基和羧基之间的氢键作用,氨基酸链在空间中旋转形成螺旋状。
- β-折叠(Beta-sheets): β-折叠是蛋白质的另一种常见二级结构,由两个或更多的平行或反平行的β链通过氢键连接在一起形成。
蛋白质结构预测在计算生物学中是一个重要且具有挑战性的问题。许多不同的技术和方法已经被开发出来解决这个问题,包括模板匹配、同源建模,以及更先进的方法,如深度学习。深度学习的方法,例如AlphaFold,已经在CASB (Critical Assessment of protein Structure Prediction)竞赛中取得了显著的成果,显示出其在预测蛋白质结构方面的潜力。
Methods for Protein Structure Prediction
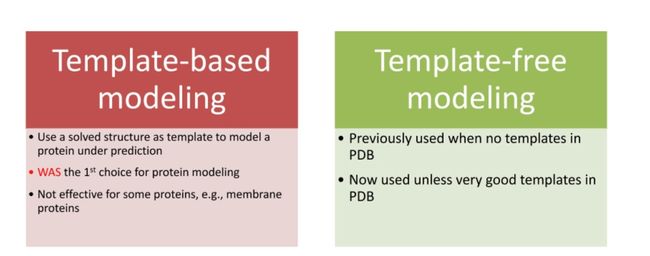
模板依赖建模和模板自由建模是两种常用的蛋白质结构预测方法。
- 模板依赖建模(Template-based modeling, TBM):这种方法依赖于已解决的蛋白质结构数据库(如PDB)中的已知蛋白质结构,将其作为模板。如果一个新的蛋白质序列与已知结构的蛋白质序列高度相似(同源),那么就可以使用这个已知的结构作为模板,预测新的蛋白质的结构。然而,对于一些没有相似模板的蛋白质,例如某些膜蛋白,此方法可能效果不佳。
- 模板自由建模(Template-free modeling, also known as ab-initio or de novo modeling):这种方法不依赖于已知的蛋白质结构模板,而是通过理论模型和计算方法预测蛋白质的结构。例如,这可能包括对蛋白质的物理和化学性质的模拟,如力学模型和电磁模型,或者使用蒙特卡洛方法进行随机搜索。然而,这种方法的计算复杂性通常较高,且预测准确性可能不如模板依赖建模。
最近,深度学习方法(如AlphaFold)已经在蛋白质结构预测中显示出很大的潜力,这种方法既可以利用已知的蛋白质结构信息(如果可用),也可以从头开始预测蛋白质的结构,从而结合了模板依赖和模板自由建模的优点
Old method: fragment assembly
以前使用超级计算机去模拟,需要很大的算力而且成功率很低

Fragment assembly是一种传统的蛋白质结构预测方法,具体过程如下:
1. 目标序列:首先,我们有一个需要预测结构的蛋白质目标序列。
2. 片段库 (Fragment Library):通过从已知结构的蛋白质中提取短序列片段来创建一个片段库。这些片段的长度通常为3到9个氨基酸。
3. Profile-Profile Alignment:然后,将目标序列的氨基酸剖面与库中的每个片段剖面进行对比。
4. 蒙特卡洛片段装配 (Monte Carlo Fragment Assembly):通过蒙特卡洛方法从片段库中随机选取片段,并将它们拼接在一起以构建蛋白质的初步模型。通过重复这个过程,产生大量的蛋白质模型。
5. 基于知识的势能 (Knowledge-based potentials):使用基于统计的得分函数(如Rosetta的得分函数)来评估和排列这些模型,选取得分最高的模型作为最佳模型。
6. 基于物理的原子精修 (Physics-based atomic refinement):最后,基于物理的能量最小化过程对选定的模型进行精修,包括优化氨基酸侧链位置和小幅度调整蛋白质的主链。
这种方法的一个主要问题是计算效率较低,且准确度受到已知结构片段库的限制。
New Strategy

这个新策略包含了以下步骤:
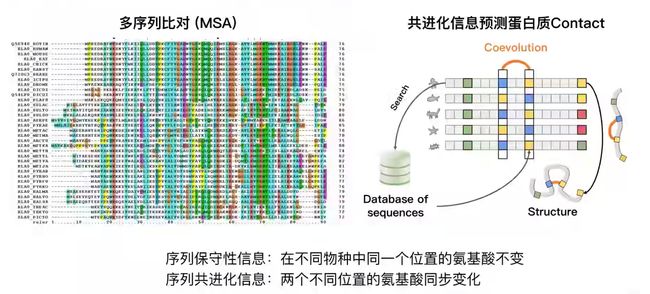
- 多序列比对(Multiple Sequence Alignment):多序列比对是一种基于序列比对的方法,它用于确定一组蛋白质序列或者核酸序列之间的相似性。通过比对多个序列,我们可以找出共享的进化保守区域,即那些在进化过程中保持不变的区域。这些保守区域通常对于蛋白质的功能和结构至关重要。
- 共演化分析(Co-evolution Analysis):这是一种寻找蛋白质内部氨基酸间联系的方法,即某个氨基酸位置的变化可能会引起另一个氨基酸位置的变化。这种共演化信息可用于预测蛋白质的三维结构,因为在蛋白质中,共演化的氨基酸对往往在空间中靠近。
- 深度神经网络预测相互作用矩阵:输入的数据包括上述的多序列比对和共演化分析结果,以及其他可能的蛋白质序列信息。这个深度神经网络的目标是预测一个相互作用矩阵,其中每个元素表示蛋白质序列中两个氨基酸在空间中的相互作用强度。
- 深度神经网络预测局部结构:类似地,也可以使用深度神经网络预测蛋白质的局部结构信息,比如二级结构(alpha螺旋,beta折叠等)和每个氨基酸的位置。
- 最小化分子力场(Minimization Molecular Force field):在获取预测的相互作用矩阵(二维)和局部结构信息(线性)后,可以使用分子模拟方法(如力场最小化或分子动力学模拟)来生成蛋白质的三维结构。这一步的目标是找到一个蛋白质结构,该结构最好地满足预测的相互作用和局部结构信息。
这个策略提供了一种有效的方法来预测蛋白质结构,与传统的方法相比,它更多地利用了深度学习和序列演化信息,因此通常可以得到更准确的预测结果。
Co-evolution Analysis

蛋白质的结构和功能严重依赖于其氨基酸的排列顺序和化学性质。如果一个氨基酸突变,使得其边链增大,这可能会干扰蛋白质的结构或者影响其功能,因为这可能会使其与邻近氨基酸的相互作用发生改变。
但是,如果另一个邻近的氨基酸同时发生突变,使其变小,这就可能可以平衡边链的大小变化,保持蛋白质的稳定性。这种情况下,两个氨基酸就会表现出共演化的特性,即它们的变化是协调的。
这种共演化现象可以帮助我们理解蛋白质的三维结构和功能,因为共演化的氨基酸对通常在三维结构中紧密接触,共同参与形成蛋白质的活性位点或者结构域。因此,通过分析多个蛋白质序列的共演化模式,我们可以预测蛋白质的结构或者功能位点。
共演化的概念可能比较抽象,我们通过一个简单的例子来理解它。
假设我们有一种具有三个氨基酸的极简单的生物体。这三个氨基酸排列在一条链上,形成了一种蛋白质。我们将这三个氨基酸分别命名为A,B,C。
在一种理想的环境下,这三个氨基酸的理想形态分别为大,中,小。也就是说,最好的蛋白质形式是大的A,中的B,小的C。这种组合能让生物体达到最好的适应环境的效果。
然而,生物体在演化过程中会发生突变,氨基酸可能会改变形态。比如,A可能突变为中型,或者B突变为大型。
现在,如果A突变为中型,那么与A相邻的B就会面临压力,因为它现在与A一样大了。为了维持蛋白质的最优形态,B可能也会随之突变为小型。这样,A和B就通过共同适应环境压力而发生了共演化。
如果我们只看一个生物体,可能很难观察到这种共演化的现象。但如果我们观察很多具有相似蛋白质的生物体,我们就可以看到某些氨基酸位置上的突变是有关联的。例如,我们可能会观察到,每当A变为中型时,B也往往变为小型。
这就是共演化的基本概念。在实际的生物体和蛋白质中,情况会复杂得多,因为一个蛋白质可能由数百或数千个氨基酸组成,每个氨基酸可能以20种不同的形式出现。但即使在这种复杂的环境中,我们仍然可以通过统计分析发现共演化的模式,从而预测蛋白质的结构或功能。
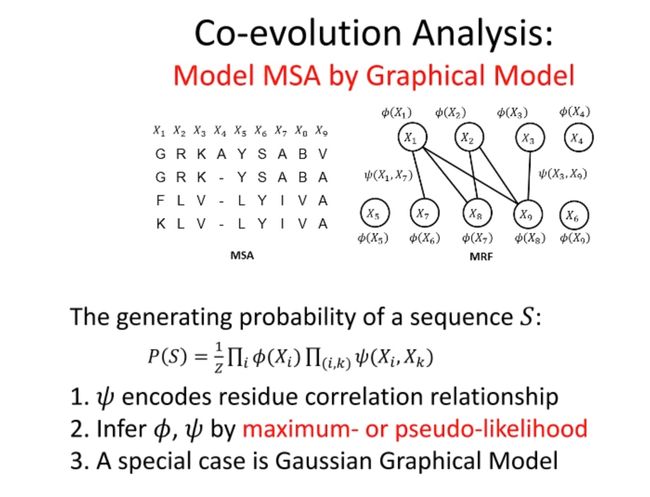
这里是对多序列比对(MSA,Multiple Sequence Alignment)的共演化分析的介绍。共演化分析在生物信息学中被广泛应用,尤其是在蛋白质结构预测和功能位点预测等领域。
这里的图形模型(也被称为图模型或马尔可夫随机场,MRF)是用来建模MSA的。它将氨基酸序列中的每个位置看作一个随机变量,并假定两个位置之间的关系可以由一个权重函数来描述。
对于给定的氨基酸序列S,其生成概率 P ( S ) P(S) P(S)可以由这个图模型来计算,其中 φ ( x ) φ(x) φ(x)表示序列中某个位置的单点概率,而 w ( x i , x j ) w(x_i, x_j) w(xi,xj)表示序列中两个位置的相关性或协变性。
这个模型的参数 φ φ φ和 w w w可以通过最大似然法或伪似然法进行估计。这种方法的一个特例是高斯图形模型,它假定随机变量服从多元正态分布。
总的来说,这个方法的目标是通过分析蛋白质序列的共演化模式,来预测蛋白质的结构或功能位点。
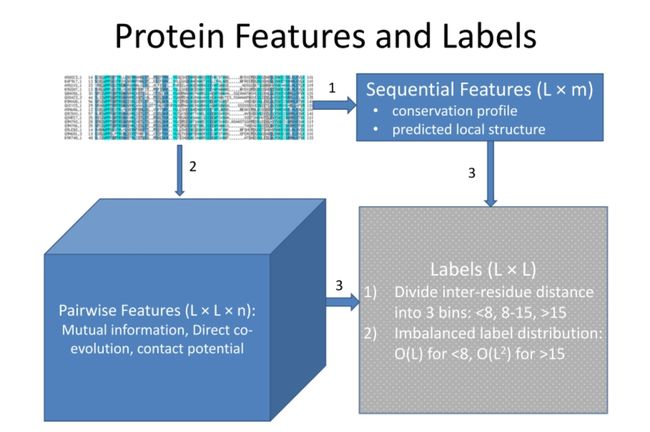
在蛋白质结构预测中,通常需要收集并利用一些蛋白质的特征和标签。这些特征和标签可以帮助我们理解蛋白质的性质并预测其未来的行为。
- 序列特征(Sequential Features): 这是指蛋白质序列中的信息,如保守性分布(conservation profile)和预测的局部结构(predicted local structure)。保守性是指某个位点的氨基酸在不同物种或不同蛋白质家族中的变异程度。预测的局部结构是指蛋白质中每个氨基酸可能形成的二级结构类型(如α-螺旋,β-折叠)。
- 配对特征(Pairwise Features): 这是指基于两个氨基酸之间关系的特征,包括相互信息(mutual information),直接共演化(direct co-evolution),接触势(contact potential)等。相互信息和共演化是量化两个位点氨基酸变化依赖性的方法,接触势则是根据氨基酸类型预测它们之间可能发生物理接触的概率。
- 标签(Labels): 这是我们想要预测的目标,例如氨基酸间的距离。这个距离通常被分为几个区间,比如小于8埃,8-15埃,大于15埃。值得注意的是,这种标签分布通常是不平衡的,因为相距较远的氨基酸对(大于15埃)和相距较近的氨基酸对(小于8埃)在蛋白质中的数量通常要多于中间距离的氨基酸对。
在收集了这些特征和标签后,我们就可以使用一些机器学习或深度学习的方法来训练一个模型,该模型可以从这些特征中学习并预测蛋白质的结构或者行为。
Towards An End-to-End Workflow
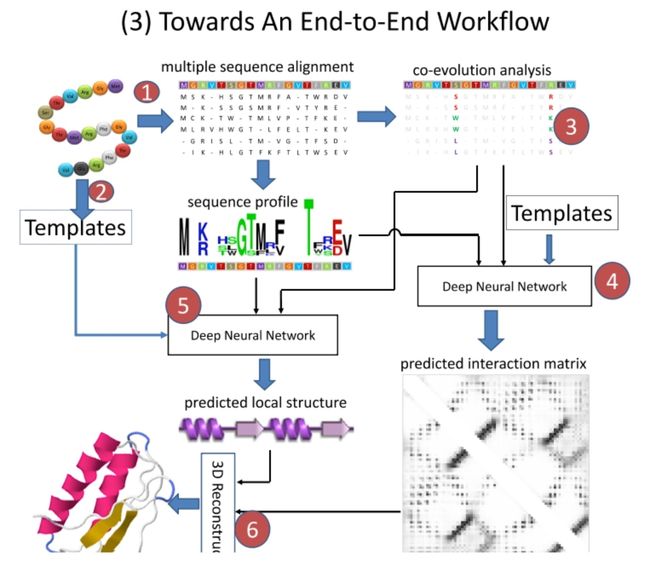
"Towards An End-to-End Workflow"工作流程引入了一个额外的步骤,即"Templates"步骤。
Templates步骤通常涉及到使用已知的蛋白质结构作为模板,这些模板在某种程度上与目标蛋白质的序列相似。这些模板可以提供额外的结构信息,有助于提高预测的准确性。这种方法尤其对于那些能找到良好模板的蛋白质序列特别有用。
在这个工作流程中,模板不仅被用于初始化预测,还被用于训练深度神经网络。这意味着,网络不仅从多序列比对和共演化分析中学习信息,而且还从模板中学习结构信息。这种结合使用数据驱动和知识驱动方法的策略有可能提高预测的准确性和鲁棒性。
需要注意的是,模板引导的预测可能对模板的质量和与目标序列的相似度高度敏感,而且不适用于那些无法找到合适模板的蛋白质。此外,这种方法可能需要更复杂的训练步骤和更多的计算资源。
AlphaFold2 architecture
补充两篇文献:
蛋白质界的 ChatGPT:AlphaFold1AlphaFold2成功秘诀:注意力机制取代卷积网络,预测准确性提升超30%


AlphaFold2是一种革新性的蛋白质结构预测方法,由DeepMind公司开发。它在2020年的蛋白质结构预测竞赛(CASP14)中表现出色,标志着蛋白质折叠问题的一个重大突破。
这种方法是端到端的,这意味着它接受一系列氨基酸(即蛋白质序列)作为输入,并输出预测的蛋白质三维结构。这与许多早期的方法不同,这些方法通常需要一系列中间步骤和手动调整。
在AlphaFold2的架构中,首先执行多序列比对,并搜索模板。然后,这些信息被喂入一个类似于Transformer的深度网络中。这个网络包括多个模块,如Evotransformer模块和Structure模块,这些模块通过大量的注意力机制和残差连接来处理序列和模板信息。
网络的输出是一个三维表示,描述了每个氨基酸在空间中的预期位置。这个表示随后被用来重构蛋白质的三维结构。
AlphaFold2的一大创新是引入了模板和多序列比对信息,这些信息通过一个强大的Transformer-like网络进行处理。这允许网络学习复杂的序列-结构关系,并提高预测的准确性。
然而,虽然AlphaFold2取得了显著的成就,但仍有一些挑战。例如,对大蛋白质的预测,以及对蛋白质动态和蛋白质-蛋白质相互作用的预测,仍然是困难的问题。
图神经网络的关键就是学习各个残基之间的关系,完成某些三维结构中每个残基的嵌入,预测最后的三维结构
在蛋白质结构预测的情况下,GNN可以被用来模拟蛋白质折叠的过程,其中节点代表氨基酸,边代表氨基酸之间的相互作用。GNN可以捕获蛋白质的局部和全局结构特征,以及氨基酸之间的复杂相互作用。
例如,可以使用GNN来预测氨基酸之间的距离,这是蛋白质三维结构预测的一个关键步骤。GNN可以通过在蛋白质图中传播信息,来捕获氨基酸之间的长距离依赖性。
补充:跟李沐精读论文
李沐老师b站:AlphaFold 2 论文精读【论文精读】

三个部分,第一个部分是抽取特征,第二个是encode,第三个是decode

- 对于第一部分,总共是以下几种输入
- 直接导入该序列
- MSA:在基因库中搜索,找相似蛋白质序 列,然后形成一个MSA(Multi sequence alignment),也叫做多序列比对。(MSA的作用是为了提取出一个蛋白质序列在多物种中的共进化信息)
- 氨基酸之间的关系,我们知道蛋白质能卷起来,就是氨基酸之间的关系,所以有一个输入的数据,是存储该蛋白质序列中,氨基酸之间的关系的(这里不是氨基酸间的空间距离,因为我们还不知道,是一些其他方面的特征)
- 最后还有一些额外特征:在结构数据库里搜,因为我们已经知道一些蛋白质的结构,然后在其中搜索得到氨基酸之间的空间距离之类的信息,得到很多模版
- 所以抽取特征主要得到两大类特征:第一类是不同序列之间的特征,第二类是氨基酸之间的特征。这两类特征再拼上一些别的东西,就可以输入编码器了

-
对于第二部分,是输入两个三维的张量
- MSA:大小为(s,r,c)
- s表示有s个蛋白质,第1个是我们要预测的人类的蛋白质,后面s-1个是从数据库中匹配来的蛋白质。
- r表示蛋白质中有r个氨基酸(多序列比对的结果,会用_补齐空缺,最后的结果应该是同长的)
- c表示每个氨基酸表示成长为c的向量(对于image就是每个像素的通道数,对于句子来说就是每个词的嵌入长度)
- Pair氨基酸对:大小为(r,r,c)
- r,r就是氨基酸个数,c就是用长为c的向量来表示一个氨基酸的特征
- MSA:大小为(s,r,c)
-
然后将两个输入进Evoformer(可以看做transformer的变种)
- 与transformer有两个不一样的地方:1.不再是一个序列的关系(比如句子),现在是一个二维之间的关系(不同蛋白质序列、同一氨基酸位点)2. 输入的是两个不同的张量,我们要融合起来
- 其他大部分就一样了。猜到可能用了transformer蛋白质3D结构也是氨基酸相互之间关系造成的,而且序列位置近的和序列位置远的都可能起重要作用,有attention 的味道了

- 然后我们得到了编码器的输出,包括要预测的人类氨基酸的所有的特征表示,还有氨基酸之间的相关信息。根据相关信息来预测每一个氨基酸的位置,最后得到我们的输出

这里还有一个回收机制,将编码器和解码器的输出,又拿回去做了编码器的输入。有点像RNN,可以看做变成了一个四倍更深的一个网络,达到更好的进度。
一些不同:每次复制的权重还是基于前面的,还有做回收的时候梯度是不反传的
暂停来自视频23:05,编码器和解码器的内部结构以后再来补充
-
编码器
- MSA按行和按列进行信息上的融合、氨基酸对的信息,相互融合学习
- Evoformer 块包含许多新颖的基于注意力和非基于注意力的组件。 Evoformer 模块的关键创新是在 MSA 内交换信息的新机制和允许直接推理空间和进化关系的配对表示。
-
解码器
- 输入氨基酸对的信息,氨基酸序列的信息、不断调整的主干的旋转和偏移信息
-
两个格外的技术
-
Noisy Student Self-Distillation
-
一种半监督学习策略,主要用于深度学习模型的训练过程中,以提升模型的表现。这种技术的名字来源于其运作机制,其中涉及到两个关键的角色: “teacher”(教师)和 “noisy student”(带有噪声的学生)。
以下是这种技术的一种通俗的解释:
- 教师模型训练: 在开始阶段,我们训练一个称为"教师"的模型。这个模型使用标记的数据进行训练,即我们知道输入数据和正确答案的配对数据。
- 生成带噪声的学生: 在教师模型被训练好之后,我们复制一份教师模型作为"学生"。不同的是,我们会在这个学生模型的训练过程中加入一些"噪声",比如数据扩充(例如图片旋转,翻转等)或者添加dropout等。
- 学生学习: 这个带噪声的学生模型会同时学习标记的数据和未标记的数据。对于未标记的数据,我们让教师模型预测标签,学生模型根据这个预测的标签来学习。
- 学生变教师: 当学生模型训练好后,它其实通常会比原来的教师模型更强大,因为它学习了更多的数据。所以我们可以用这个学生模型来替代原来的教师模型。
- 迭代过程: 这个过程可以重复进行,每次用新的学生模型替代教师模型,并且生成新的学生模型来学习。这就是为什么这个过程被称为"自我蒸馏"的原因,因为模型在不断地学习自己的知识。
-
因为要加到氨基酸对里面 每列表示的是一个氨基酸 选择两列就是两个氨基酸的信息
摘录了一些小伙伴的b站评论:
问:首先是一个不成熟的小建议:我觉得李老师在做AI for science这类论文的解读时,要是能够对这个science任务做个简要介绍就更好了。比如这个任务是要从什么已知条件得到什么未知结果,以及目前非AI方向是用哪一些方法来完成这个任务。这应该能有助于我们更好地理解作者在这篇文章中某些做法的意图。这是我这几天找的资料整理的,生信的小伙伴们看看有没有什么问题:
蛋白质的三级结构是由一级结构决定的,每种蛋白质都有自己特定的氨基酸排列顺序,从而构成其固有的独特的三级结构。蛋白三维结构预测就是指输入一段蛋白序列(一级结构),输出蛋白所有原子的三维空间坐标。当前对蛋白质三位结构进行预测的方法,除了文中提到的Cryo-SEM,还有通过同源蛋白质的同源建模方法。具体步骤如下:首先选择最佳模板3D结构后进行序列比对,第一次的序列比对通常使用BLOcks替换矩阵执行。第二次序列比对(也称为比对校正)用于构建骨干三维结构。然后对无模板区域或者相似性比较低的区域进行loop建模,最高精度可达12~13个残基。接着是侧链重建,通过依赖主链的旋转体库进行构象搜索。接下来应该通过各种质量评估工具对结构进行改进和验证。我想在本文中输入的MSA,正是对应模版的蛋白质序列,而template也就是这些模版蛋白质序列对应的结构信息。
答1:MSA和template并不一样,MSA的序列远比template里面的序列多的多。MSA建模的思想是来自共进化分析,也就是只通过MSA完全不要序列理论上也能得到3D结构;template建模就是您说的同源建模的思想。但是AF里面保证精度的核心是MSA
答2:MSA 是在seq databse里搜索和查询目标相似的序列来提取共进化信息,也就是在序列层面上获取一些残基之间的接触关系来指导最终的结构预测,template 基于序列直接搜索同源结构,获取的是模板结构在三维空间中残基之间的相对位置信息。MSA 的适用性要更广于template,主要原因在于蛋白质序列的数据库规模要远大于解出结构的蛋白质构成的结构数据库。
补充:钟博子韬同学的报告
交大这位同学关于alphaflod的报告更加面向生物背景
- 背景
- 实验方法:低通量、高精准度。计算方法:高通量、低精准度
- 海量的序列信息,少量的结构信息。需要高精度高通量发现蛋白质结构的方法
- 预测蛋白质的contact map理论基础
- contact map可以是一条蛋白质序列中,两两氨基酸之间的距离的map。也可以是小于某个阈值,看作两个氨基酸是contact的
- 预测contact map可以更方便的迁移使用一些cv里的网络模型。比直接预测3d坐标简单

通过共进化信息,比如两个在序列上比较远的氨基酸位点,却表现出共进化现象,说明这两个位点在3d空间中可能距离更近
- 对于alphafold2来说,其主要特点
- 是一个端到端的架构,输入序列,输出是结构,不是contact map
- 1d(MSA)和2d(氨基酸之间的信息,类似contact map),二者在训练中使用Attention更新,不断交换信息
- 通过Structure Module,3D Equivariant (等变),可以直接输出三维结构

- 精彩之处
- 模型输入——更强大的MSA和Templates
- 使用Recycling进行多轮迭代训练和测试
- 基于Attention提取进化信息:按行/列提取MSA的相关性、氨基酸之间关系的信息提取,MSA和pair之间的信息交互

- 氨基上的N、中心碳原子和羧基上的碳原子,构成一个三角形,称为Residue Gas。这个三角形的形状不太会变,而绝对坐标会变。输入的是,(encoder输出的)目标蛋白序列、学习到的contact map以及初始骨架。通过IPA模块不断更新,让结构表示能成为3d坐标。最后补上侧链


- alphafold会给出自己预测的这个结构的置信度

- 回顾

-
AlphaFold的优点总结和补充
- 基于recycling的迭代优化。这一点在很多领域己经得到过应用,比如计算机视觉中的姿态估计 (post estimation)
- 广泛应用的Attention架构。将二维的表横着做Attention、再竖着做Attention,对于图可以在局部做Attention,不断精化了Embedding过程;Structure module中也继续用到了Attention
- 实现了端到端(end-to-end)架构。完整建立了用于蛋白质结构预测的端到端架构,让模型能够在提升准确度的同时,融合结构的优化步骤。
- 半监督学习拓展训练集(Self Distillation)。用带标签的数据先训练一遍,再用无标签的数据预测一遍形成新的数据集,然后再混合继续训练。这种方法曾经在Google Brain的nois student使用过,在这里再次得到了应用
-
蛋白质结构预测的本质:
- 从共进化信息推断蛋白质结构的contact
- 共进化信息并不是物理的作用关系
-
AlphaFold通过单体的共进化信息,来预测结构之间的信息(三级结构),这套理论还不能迁移到复合物的预测中(四级结构)
-

Multimer模型对输入进行改进
- 所以后来的工作方向是要预测复合物的结构

预测结果
-
精确度的显著提高
-
Recycling的必要性:有的蛋白很快就能够折叠,有的蛋白很慢
- 多轮迭代优化有一定的必要性,较为复杂的蛋白可能在优化流程最后
(4轮优化)才能折叠到正确的结构
- 多轮迭代优化有一定的必要性,较为复杂的蛋白可能在优化流程最后
-
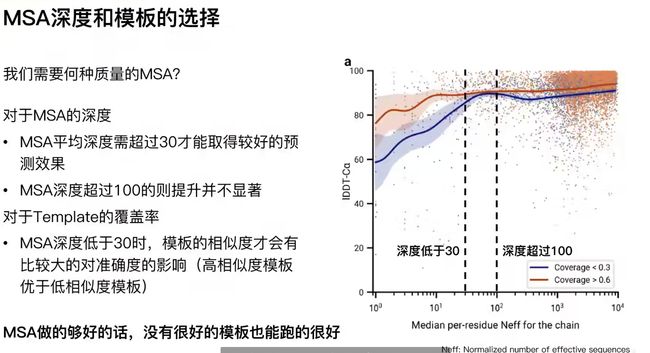
MSA深度和模板的选择
- msa做的够好的话,没有模版区别不大
- 如果msa做的很差,一般也找不到模版,除非做实验,这个也要改代码,以后可能会优化
- 不要对alphafold做模版,超过30的msa在bfd中很好找
-
-

-
评价指标

- 例子
