智能合成:有机合成中的智能化与自动化
文章的内容来源于书籍《分子科学中的机器学习》,全书的获取方式在文章结尾。
1. 摘要
在本章中,我们将简要介绍有机合成中的智能化和自动化。本章包括五个主要部分。在第一部分中,讨论了计算机辅助的反综合规划的例子。在第二部分中,将讨论反应结果预测模型,它利用机器学习来建立反应产率、反应选择性、反应中间体、反应类型、反应产物和反应条件预测的模型,还简要介绍了建立反应预测模型的基本要素。第三部分将介绍机器学习辅助反应发现和优化的相关例子,然后是自动合成和实验室机器人的特殊代表性案例。最后,讨论化学家可能关心的机器学习和自动化的几个问题。
自从1828年合成了第一个由无机化合物尿素制成的有机分子[1]以来,有机化学一直出于一种低效和劳动密集型过程。过去,合成化学家需要几个月或几年的时间来发现新的反应和优化反应条件。得力于互联技术引发的第四次工业革命(工业4.0),有机合成也进入了合成4.0,这是一个将机器、自动化和人工智能(AI)统一起来的新时代,可以为化学合成提供智能解决方案。合成化学家不仅可以利用高通量实验(HTE)进行快速多反应参数筛选、并行合成大量化合物的[2-14]。也可以利用合成自动化和实验室机器人技术,使他们从日常的合成工作中解放出来[15-32]。并且,利用大数据和机器学习(ML)[33],有机反应正在变得可预测,反应的发现将不再只是通过直觉或偶然发现[26,30,31,34-38]。有机合成现在正走在数字化和自动化的高速公路上。
2. 逆向综合规划
自1990年诺贝尔化学奖获得者E.J.科里在20世纪60年代构思并正式确定以来,有机化学家和计算机科学家一直在探索计算机辅助(追溯)合成计划的道路[41]。
20世纪80年代至90年代,在逻辑和启发式方法应用于合成分析(LHASA)十年后,第一个合成路线规划的计算方法被开发出来[48],之后合成设计和反应预测的新算法迅速出现。例如,CAMEO被开发用于通过分析亲核和亲电位置来预测反应[49,50]。EROS能够根据键极性、电负性和共振效应识别用于回合的先导结构[51]。SOPHIA 能够用最少的用户输入来预测反应结果[52,53],KOSP可以用来预测逆合成靶标[54]。但这些预测工具存在明显的缺点,如适用范围较窄。
2017年,Segler和他的同事报告了一种知识图谱方法,该方法使用半反应的概念预测可能的产物[55]。随后,他们又报告了一种用于逆合成和反应预测的混合模板和神经符号的方法[56],该算法可以模拟人类的决策方式,避免反应性冲突。与基于规则的专家系统相比,该方法的速度提高了150倍。然而,这个系统没有考虑到立体化学。2018年,他们结合蒙特卡洛树搜索(MCTS)算法用于发现了有机小分子的逆合成路线[57]。MCTS与指导搜索的扩展策略网络和过滤网络相结合,可以预先选择最有希望的逆合成步骤。在双盲AB测试中,人工智能生成的合成路线被认为与人类化学家报告的路线相当。然而,对于复杂天然产物的逆合成和定量预测异构体间的比率仍然具有挑战性。
2019年,Coley等人基于他们之前的工作[58]提出了一种计算机增强化学合成的策略,其合成路线是从数百万份化学出版物中挑选出来的,然后在计算机中进行模拟,以最大限度地提高成功的可能性,最后由模块化的有机械臂的自动合成平台执行(方案8.1)[59]。15种药物或类药物化合物的合成证明了这一发展战略是可行的。

方案 8.1 Coley 等人的计算机增强化学合成策略
2020年,GrzyBowski等人在他们早期研究的基础上开发了一个逆合成程序Chematica(或Synthia)[61]。该程序在决策中嵌入有机化学知识,增强结果的因果关系,这使得它能够在多步合成中进行“战略制定”。程序设计的三条复杂天然产物的合成路线在实验室中得到了成功的验证,展示了这种专家级自动合成规划程序在有机合成,特别是全合成中的潜力。
上面所述的逆向合成规划方法都基于模板的。这些模板库需要人工编码,这是非常耗时、且可能存在预测不切实际[62]和反应知识过时[42,63,64]的问题。从反应数据集中自动提取转换是手动编码反应规则的替代方案[65-67]。例如,GrzyBowski等人通过对Beilstein数据库中的所有反应进行编码,创建了一个“有机化学网络”[68]。通过搜索这个网络,可以成功地绘制出与该数据库中已有分子足够相似的任何分子的合成路径[69]。不幸的是,该算法仍然缺乏预测未知反应结果的能力[70,71]。此外,由于人类的偏见和不完整性,从文献中收集的这种合成数据可能具有误导性,例如,缺乏失败的结果[72,73]。
鉴于基于规则或模板库的逆向综合规划方法的缺点,专家们开始开发无模板方法。例如,Liu等人报告了一个用于逆合成预测的无模板数据驱动的神经序列到序列(Seq2seq)模型[77]。这个端到端的训练模型使用SMILES作为输入和输出,具有由两个递归神经网络(RNN)组成的编解码器体系结构,这在前向反应预测任务中得到了成功的演示[78-80]。金等人报道了一种探索产物分子空间的有效方法,该方法利用图形卷积神经网络(GCNN)来确定反应中心,而不是通过反应模板生成候选产物[81]。反应中心识别符和候选排序模型都是从Weisfeeller-Lehman Network(WLN)及其变体,如Weisfeeller-Lehman Difference Network(WLDN)[82]构建的。尽管SMILES 被广泛地用来代表分子,但SMILES 的语法是如此脆弱和复杂,即使是一个字符的变化也足以使整个SMILES 串失效。最近,Lee的团队使用Transformer体系结构[83,84,85,86],开发了一个新的无模板逆合成预测模型RetroTRAE。与其他基于SMILES 的Transformer模型相比,该方法表现出了类似的性能[87-94]。
除了常规有机分子的合成外,化学家们还使用规划算法来合成多肽。由于肽设计传统上是由人类专业知识和直觉指导的,这很难扩展,而且容易受到人类偏见的影响,最近,Fry、Sankaranarayanan和他们的同事记录了一种机器学习工作流程,将MCTS和随机森林(RF)与分子动力学模拟相结合,开发了一个完全自主的计算搜索引擎,以发现具有高自组装潜力的肽序列[95]。该工作流被称为AI-Expert,能够高效地搜索大空间的三肽和五肽,具有比人类专家更好的预测能力。通过AI-Expert获得了具有高自组装倾向的非直观序列,这表明其具有克服人类偏见和加速肽发现的潜力。然而,AI-Expert没有收到任何来自实际实验的反馈,并且在计算分数和实验分数之间只发现了适度的相关性。此外,这位人工智能专家似乎也有自己的偏见:对脯氨酸的偏爱和缬氨酸的有限倾向。
3. 反应预测
除了计算机辅助的逆合成规划外,使用人工智能来预测反应结果,如给定反应物的反应产物、产率和选择性,也很有吸引力。实现反应预测的目标有三个关键部分(图8.1):为模型训练提供足够的高质量数据集,适当的描述符来描述反应变量,以及建立预测模型的拟合算法。
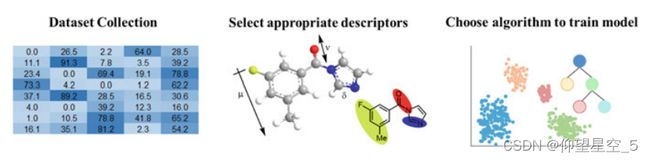
图8.1建立反应结果预测模型的步骤
训练数据可以从互联网上获得,如SciFinder、Reaxys®、专利、已出版的文献、专有数据库或从出版物中提取[96-98]。然而,这种数据的质量可能不可靠,并且可能省略诸如温度之类的原始数据或诸如立体化学之类的关键信息。数据不平衡是另一个问题,因为当化学家发表他们的研究成果时,负面结果总是被丢弃[99-101]。获取所需数据的另一种途径是内部实验。为了生成大数据集(至少数百个数据点),需要hte。HTE是一种在小规模上筛选化学实验的技术。通常,HTE指的是小型化井板批次格式,包括24井、96井以及384井阵列,而需要专门设备的UltraHTE指的是1536个或更多阵列。在进行反应时,可以通过单通道或多通道手动微吸管或商用液体处理自动化系统处理小体积的溶剂或液体试剂,如ChemSpeeSwing[104,105]、Tecan Freedom EVO[5]、Labcell Echo[106,107]、Thermo Matrix[108]、SPT LabTech Modio[102,108-111]和Unchaated Labs Junior[5]。另一方面,由于性质的多样性,固体分配在HTE[112]中是有问题的。其中一个解决方案是在微小的玻璃珠表面均匀地涂覆固体化学物质[113,114]。然而,我们的内部实践表明,一些固体化学品不适用于“ChemBeads”战略,因为固体不能均匀地覆盖在玻璃珠上。已记录了许多使用热传导技术进行条件筛选或数据收集的例子[102、108、110、115-119]。对于那些对HTE感兴趣的人,可以参考相应的综合评论[2-14]。
分子描述符用来表征分子。这样的描述符由算法生成,作为分子属性的数学表示[120],根据它们的“维度”[121]。一维(1D)描述符包含大量的物理化学性质参数,如分子量、对数P、极性表面积等。二维(2D)描述符包括从分子的2D表示导出的结构片段或连接性指数。三维(3D)描述符,例如分子形状,是从3D分子结构导出的。不同类型的分子指纹,即与分子性质相对应的数字或浮点数的矢量,被用作反应预测算法的描述符。摩根分子指纹图谱(MMF)[122]和扩展环形指纹图谱(ECFP)[123]已被广泛用于预测分子性质,包括HOMO-LUMO间隙[124]、蛋白质-配体结合亲和力[125]、药物毒性水平[126],以及预测合成可及性[127]。已经报道了指纹识别方法的大量发展[128-135]。实际上,人们可以使用RDKit或Mordred[136]这样的软件库将给定的分子转换为一维或二维描述符。神经网络的使用可以为从2D分子结构[75,129,134,135]的反应预测模型提供分子图表示。密度泛函理论(DFT)计算和分子力学(MM)计算被用来产生物理意义的描述符,这些描述符表示三维空间[137]中的电子和空间性质,包括HOMO-LUMO能量、扭角、空间位阻参数[138,139]和埋藏体积[140,141]。据报道,有几次尝试通过反应能垒来预测反应的可能性[142-147]。尽管与启发式方法相比,量子计算可以将探索引导到更广泛的反应范围,但对于每个额外的反应族,新的计算将花费大量的时间和金钱。Baldi和他的同事记录了通过反应机制观点预测反应[148-150]。他们使用Reaction Predictor算法来识别分子的潜在电子源和电子汇,列举并对可能的相互作用进行排序[64,151]。然而,手动编码机械性规则将是有问题的,并限制可伸缩性。江、罗和他的同事最近设计了一个化学信息分子图(CIMG)来描述化学反应。化学信息包括核磁共振化学位移作为顶点特征,键离解能作为边特征,催化剂/溶剂信息作为全局特征,用于表示分子和反应。结合图神经网络(GNN)模型和MCTS,有效地预测了具有推荐催化剂/溶剂的全多步逆合成规划路线。
有了高质量的数据集和适当的描述符,确定拟合算法是构建反应预测模型的最后一步。主要是将给定的数据集分为两个子集:用于匹配反应算法的训练数据集和用于验证训练模型的测试数据集。在某些情况下,数据集将被划分为训练集(以初始地拟合模型)、验证集(以在调整模型的超参数的同时提供对模型的无偏评估)和测试集(提供对模型的无偏评估的不可见的数据集)。通常,将针对给定的数据集测试和调整多种算法,以找到性能最佳的算法。为了测试训练模型的稳健性,通常执行外部验证,这意味着在新的样本外数据集中测试模型。评估预测模型的性能有几个度量标准。简单地说,精度用于评估分类模型(例如,用于预测是否会发生反应),而R2值(也称为决定系数)和均方根误差(RMSE)通常用于评估回归模型(例如,用于预测反应产率)。精度和R2总是较大,而RMSE较低是首选。
过去,都认为用来训练算法的数据集越大,模型的性能就越好。然而,影响反应预测模型性能的是数据集的质量,而不是数据集的数量。几个例子已经证明,如果利用适当的算法,可以使用较小的高质量数据集来获得良好的性能预测模型[153-156]。
一般来说,反应预测模型有两个主要目的。一种是预测给定条件下的反应产率或产物的区域/立体/对映体选择性。另一种是使用算法来推荐转化一组给定反应物的条件。
3.1 反应产率预测
2018年报道了一个使用ML预测反应产率的最近的例子,Doyle和他的同事描述了如何使用通过[109]获得的数据在多维化学空间中利用机器学习来预测Pd催化的芳基卤化物与4-甲基苯胺的Buchwald-Hartwig反应的反应产率。他们在存在各种潜在抑制添加剂的情况下,为反应组分创建了原子、分子和振动(反应条件?)描述符。以这些描述符为输入,以反应产率为输出,结果表明,RF算法的预测性能明显优于线性回归分析。RF模型也被成功地应用于稀疏训练集和样本外预测,这表明它有可能促进综合方法学的采用。然后进一步应用相应的方案来寻找不同类别的醇与不同的磺酰氟支架在脱氧氟化中的最佳条件[155]。然而,这些算法不能应用于超出训练集中数据范围的预测。
深度学习[157,158]技术已被应用于许多反应预测任务。例如,Liao和他的同事构建了一个基于深度学习的模型,用于预测以二氧化碳为无迹导向基团的邻烷基芳基酮受阻的间位C-H键的新型选择性官能化反应产率(方案8.2)[159]。这种一锅三步的转化包括光诱导的Csp3-H羧化,芳基乙酸催化的受阻的间位C-H键功能化,以及微波辅助的脱羧基。在这项工作中,HTE被用于条件筛选、底物范围探索和机器学习的实验数据条件。基于通信消息传递神经网络(CMPNN)结构,他们开发了一个模型,即用于反应产率预测的通信消息传递神经网络(CMPRY),以预测该串联反应的反应产率。该模型能很好地预测一系列未知底物的反应产率,其R2值为0.706,平均绝对误差为8.4%。
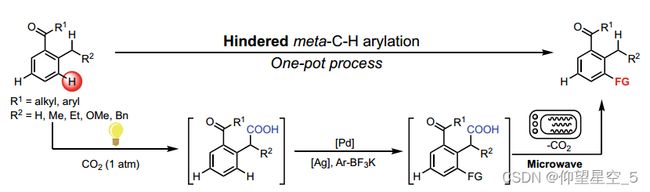
方案8.2邻烷基芳基酮受阻间位C-H键的选择性官能化
最近,廖的团队利用AutoGluon[160],一个开源的自动机器学习框架,建立了一个针对TEMPO介导的伯醇氧化的产率预测模型(方案8.3a)[161a]。通过hte访问的3444个实验数据点,他们使用AutoGluon自动训练、调整和集成了几种不同的模型,包括神经网络、LightGBM Boost树、CatBoost Boost树、随机森林、极端随机树和k近邻。在这种多标签策略中,AGMP首先将反应分类为反应性(Y)或非反应性(N),然后将预测的产率输出为回归结果,并将生成的标签“Y/N”作为附加描述符。通过结合HTE和ML,他们还报道了Ir(I)催化的羧酸和亚砜叶立德的高选择性O-H键插入反应(方案8.3b)[161b]和1,4-二氢吡啶(DHP)衍生物催化的脱羧化硒反应(方案8.3c)[161c]的反应产率预测模型。
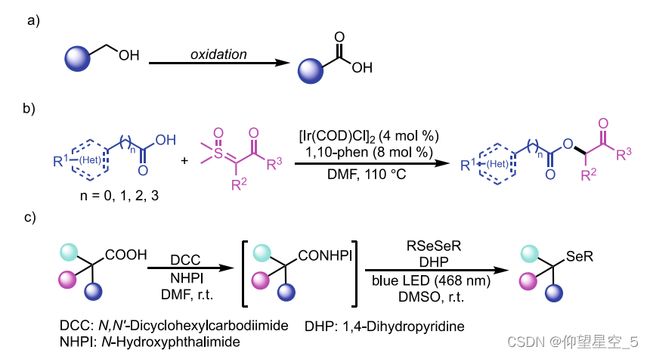
方案8.3廖氏小组预测模型的反应方案
3.2 反应选择性预测
2019年,Sigman和他的同事报告了一个预测手性磷酸催化的亚胺亲核加成反应对映体选择性的多元线性回归模型[162]。每个异构体(E-亚胺或Z-亚胺)的过渡态之间的活化自由能差G-‡值被指定为一个综合的多元线性回归模型的输出。随后将数据按过渡态几何划分,以建立两个有重点的多元线性回归模型。样本外预测揭示了该对映体选择性模型能够预测不同反应和新型催化剂结构的结果。
Denmark等人通过机器学习开发了一种化学信息学指导的手性催化剂选择工作流程[137]。他们首先构建了一组BINOL衍生的手性磷酸化合物物库,并计算了每个手性磷酸支架的相关化学描述符。发明了一种称为平均空间占有率(ASO)的新描述符来描述支架结构周围的立体环境。然后选择催化剂的代表性子集作为通用训练集(UTS),其与反应机理无关,并且可用于优化由这种支架催化的任何反应。遵循训练数据收集,机器学习方法,包括支持向量机。和深度前馈神经网络,建立了对映体选择性预测模型。一组外部催化剂被用来评估这些模型。一系列手性磷酸催化的硫醇加成反应证实了这些模型的准确预测能力,表明它们可用于手性催化剂的选择和对映体选择性的预测。
建立了区域选择性的预测模型。传统上,区域选择性基本上是由化学家的直觉预测的[163]。然后使用Harmmett[164]和Taft参数[165]以及计算的核磁共振位移[166]来近似计算芳香族核心的亲核性。利用密度泛函[167]和半经验量子力学(SQM)[168,169]对亲电取代芳香族化合物的区域选择性进行了较好的预测。2020年,Jensen等人提出了一种多任务方法来研究亲电芳香取代反应的位置选择性[170]。基于WLN结构,建立了127种反应类型的亲电芳烃取代反应的中心选择性预测模型,总体Top-1准确率为84%。利用量子力学描述符,他们最近开发了一个预测平台,用于预测芳香C-H功能化的区域选择性[171]。
Hong和他的同事开发了一个快速而可靠的模型,用于预测自由基C-H官能化对杂环的区域选择性[172]。DFT用于生成分子的物理有机特征,而RF用于提供满意的回归模型。样本外验证的准确率达到94.2%。
最近,Hartwig的小组报告了一个混合计算模型来预测Ir催化的复杂分子中Csp2-H键硼基化的位置选择性[173]。建立预测模型(SOBO)涉及DFT、SQM、化学信息学、线性回归和最大似然方法。与另一种预测模型(即WLN)和人类直觉相比,SoBo在完全准确地命中样本外验证集的确切位置方面表现出更好的性能(图8.2)。

图8.2人工和模型位置选择性预测的比较。正常化满分为100分
3.3 反应中间体、反应类型、反应产物和反应条件预测
ML也可以用来建议给定反应物的反应中间体或产物。2015年,Kabushv和Ley等人报告了一个新型的基于网络的集成系统,该系统可以计算、可视化和存储来自开源组件的系统化的关键反应参数(图8.3)[174]。用户可以绘制2D结构,该2D结构将被处理以通过网络界面生成最稳定构象的3D结构,然后通过DFT计算为每个构象创建所有可能的中间体。然后,通过快速而全面的几何优化确定了最合理的中间体。增加了对计算的许多错误检查,并使用不同的输入选项和初始猜测重新启动,以使计算完全自动化和健壮。作为概念验证,该网络平台实现了对锂化反应的区域选择性和底物的相对反应性的合理化和预测。计算结果与实验数据吻合较好。
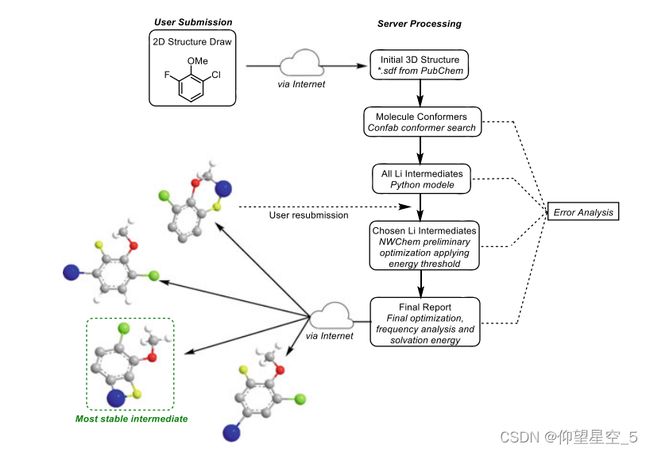
图8.3锂化反应结果预测工作流
2016年,Aspuru-Guzik的团队应用指纹方法,包括神经分子指纹,来预测给定的一组反应物和试剂的有机化学反应类型,甚至产品分子[175]。这种基于指纹的神经网络算法实现了85%的测试反应的准确率,而80%的教科书问题被正确识别。Jensen的研究小组还报告了一个关于给定反应物的反应性的GCNN模型[176]。
Jensen的团队描述了一个通过结合刚性反应模板和机器学习来预测给定一组反应物分子的主要化学反应产物的模型(方案8.4)[177]。该模型框架将基于模板的正向枚举和基于ML的候选排序相结合。应用所有反应模板来列举给定反应物的候选反应,然后对候选概率进行排序以找出最可能的产物。虽然这种两步法可以在五次交叉验证中以72%的准确率预测记录的产品,但不能在模板库范围之外进行预测。
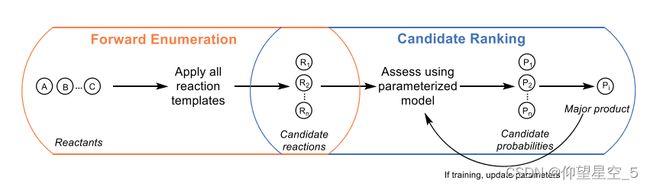
方案8.4主要产品预测的正向枚举和候选排序相结合的模型框架
Jensen的团队还开发了一个神经网络模型来预测反应。反应条件包括催化剂(S)、配体(S)、溶剂(S)、试剂(S)以及温度[58]。该模型能够预测与报告条件具有相同功能的确切条件。Varnek和他的同事也报道了预测Michael反应和保护基团去保护条件的ML模型。
3.4 其他
除反应结果外,ML还可用于物化性质预测。例如,Jiang等人使用ML预测了催化剂表面-分子吸附相互作用[180]、蛋白质的红外光谱(IR)[181]以及蛋白质二级结构的识别[182]。GrzyBowski及其同事使用GCNN预测了C-H键的pKa值[183]。莫的团队最近描述了另一个例子,他们使用自动化平台进行薄层色谱(TLC)分析,促进了在17种溶剂条件下从387种化合物中高通量收集4944个标准Rf值[184]。用ML方法建立了有机化合物结构与其极性之间的替代模型,并用延迟因子(RF)反映了它们的极性。训练好的ML模型可以根据有机化合物在不同溶剂组合中的Rf值曲线的预测,为选择净化条件提供一般性的指导。此外,该模型还可以通过预测的Rf值的差异来预测多个化合物分离的概率。
4. 反应发现和优化
4.1 反应发现
新反应的发现本质上是不可预测的、耗时且费力的。例如,在探索新反应时,初始条件通常不是最理想的,如果所需分子以痕量产物的形式出现,尤其是在微米或纳米尺度上,则很容易被忽视。在这种情况下,反卷积算法可用于突出这些痕量峰作为潜在的新颖反应产物[185, 186]。另一方面,机器学习在新化学空间导航中的应用仍未得到充分探索[187]。原因是评估未知反应与不可预测产物的反应性比使用用于优化已知目标化合物的指标(例如产率或选择性)更具挑战性[188]。
Cronin 的小组表明,由 SVM 控制的合成机器人可以显着加速有机反应的发现 [189]。液体处理机器人通过从起始材料池中选择反应物来进行反应,并通过在线分析实时监控反应结果。机器学习被用来建立化学空间的模型,并推荐下一步的实验并控制机器人。该系统不仅可以比手动更快地执行和分析反应,还可以通过基于机器学习的决策过程预测约 1000 个反应组合的反应性,准确率超过 80%。此外,化学家通过手动跟踪预测成功发现了四种反应。这项研究代表了通过由人类专家培训的人工智能驱动的化学机器人开发智能自动化方法来探索反应空间的关键一步。
最近,Zahrt 及其同事应用机器学习来指导电化学反应的发现 [190]。首先开发了一种分子表示,能够使用有限的训练数据生成通用模型,然后通过自动化实验收集电化学反应的数据。然后,如此大量的反应被分类为能力或不能力,并用于训练分类模型。通过在计算机中筛选 38,865 个潜在反应并识别许多新反应作为能力来评估该反应性预测模型,其中 80% 被发现具有反应性。
4.2 机器学习辅助反应优化
传统上,反应优化研究主要基于化学家的知识,并最终使用实验设计(DoE)方法的原理。尽管DoE可以自动化以加速预先确定的实验条件的筛选并允许响应面建模,但它需要大量的实验来拍摄最佳条件[191, 192]。尽管应用了 HTE 和综合自动化,但访问大量数据点并不是一项简单的任务。因此,在有机化学中使用机器学习是一种有吸引力的辅助策略[45]。
2018年,Aspuru-Guzik团队报告了一种基于贝叶斯优化的化学和实验背景下的全局优化算法[193]。该算法称为 Phoenics,可以根据之前所有的观察提出新的条件来定位最佳条件,从而避免冗余评估。例如,Phoenics 在 Oregonator 中展示了其适用性,这是一个描述非线性化学反应网络的复杂案例研究。尽管搜索空间很大,但 Phoenics 可以快速识别产生所需目标动态行为的条件。
Sunoj 和同事开发了一个实用的 ML 模型来发现轴向手性 BINAP 衍生催化剂家族中烯烃和亚胺不对称氢化的催化剂 [194]。利用一组 368 种底物-催化剂组合的分子参数,RF 能够做出准确的预测,与实验数据相比,预测的对映体过量 (e.e.) 的 RMSE 为 8.4 ± 1.8。该 RF 模型还可以成功预测一系列样本外数据,表明其在发现不对称催化剂以及选择特定催化剂的底物方面的潜力。
2021 年,Adams 和 Doyle 等人报告了贝叶斯反应优化模块化框架,将优化算法集成到日常实验室实践中 [195]。他们收集了钯催化直接芳基化反应的大型基准数据集,对反应优化中的算法优化与人类决策进行了系统研究,并将贝叶斯优化应用于 Mitsunobu 和脱氧氟化反应。这项工作可能有助于更有效地合成增值化合物,从而能够就进行哪些实验提供更明智的、数据驱动的决策。
2021 年,Wang 和同事利用机器学习加速了用于 CO2 还原的铜催化剂的发现和优化 [196]。这种机器学习辅助方法可以首先找出减少二氧化碳的显着特征,然后细化关键特征,然后进行催化剂设计和验证。
除了小分子和肽合成之外,机器学习还应用于材料合成的优化。特此选取两个例子。 2016 年,Norquist 和同事报告了一种借助 SVM 从失败的实验中发现机器学习辅助材料的方法(图 8.4)[197]。根据历史反应数据生成的机器学习模型用于推荐要执行的新反应,并生成关于晶体形成的人类可解释的假设。

图 8.4 机器学习辅助材料发现的示意图
Cooper 的团队将机器人实验与高通量计算相结合,探索高活性线性聚合物作为析氢光催化剂[198]。 ML 模型中使用四个变量来导航不同聚合物之间的结构-性能关系,包括预测的电子亲和力、预测的电离势、光学间隙和聚合物颗粒在溶液中的分散性。
4.3 通过 ML 和 PAT 集成进行反应自我优化
过程分析技术(PAT)[199],包括紫外可见光谱(UV-vis)[200]、红外光谱(IR)[201]、拉曼光谱(NMR)[203]、质谱光谱分析 (MS) [204]、气相色谱 (GC) [205] 和高效液相色谱 (HPLC) [204c, 206] 在连续流处理中发挥着越来越重要的作用 [19, 199b, 207],它能够对各种应用进行实时反应监测,例如动态[201e,208]和动力学[209]分析、在线手性监测[210]、过程控制[201b,211],最重要的是,自我优化[21, 212]。
2018 年,Abolhasani 的团队设计了一个自主微尺度流动化学平台,该平台配有在线 HPLC 装置、蒸发光散射检测器 (ELSD) 和 MS 模块,用于对均相 Rh 催化的烯烃加氢甲酰化进行基础和探索性研究 [204c]。单液滴管中管微反应器提供了优异的传热和传质速率,并消除了间歇式反应器的启动/关闭时间延迟。该方法允许使用少于 1 mL 的总试剂体积连续进行 43 个加氢甲酰化实验。通过该微流体平台系统地研究了配体库的催化活性、化学选择性和区域选择性。
Kappe 的团队展示了在模块化微反应器系统中集成三种不同的实时分析仪器,即在线红外和核磁共振以及在线 UPLC,用于多步有机金属反应的优化和过程中监测[213]。这种模块化流动反应器可以通过大量传感器以及集成控制系统进行实时反应监控。
在尝试优化钯催化的 Suzuki-Miyaura 反应时,Christensen 和 Jason 等人开发了一种与在线 HPLC 和 Chemspeed 液体处理机器人集成的自动动力学分析系统,以促进自动反应优化和监测 [214]。通过利用这个自动化系统,作者成功地挖掘了机理见解,并为铃木-宫浦偶联反应绘制了更有效的预催化剂。
一个更有吸引力的反应优化策略是将内联/在线分析反馈与智能算法相结合,这使得优化系统能够分析先前的反应结果,以便在短时间内适应和修改以下实验[207c,212a,215–217] 。这种“闭环”系统将通过模仿人类实验者以试错的方式自动进行化学空间探索,需要三个基本部分:执行和分析反应的机器人、解释分析数据的程序,以及将反应结果与输入和过程参数相关联的算法,该算法将为下一个反应推荐预测的最佳参数并关闭循环(图8.5)[103,188,206b,218–220]。

图8.5 “闭环”反应优化
2007 年,deMello 的团队通过应用在线荧光光谱作为荧光纳米颗粒受控合成的监测工具,开创了这一概念 [200]。几个研究小组很快开始采用这种“闭环”策略来进行反应自我优化。 Poliakoff 的团队开发了一种超临界二氧化碳 (scCO2) 自优化自动化反应器。该技术能够连续调整关键反应参数并通过在线气相色谱监测对反应结果的影响,已应用于一系列反应,例如醚化[221]、氢化[222, 223]、羟醛缩合[ 224]和甲基化[225-228]。 Poliakoff 及其同事还构建了一个自动反馈回路系统,该系统具有稳定的基于分支和拟合噪声优化 (SNOBFIT) 算法 [229],该算法基于超级修改单纯形 (SMSIM) 算法来优化 scCO2 [205] 中的反应。这种方法可以优化同一反应混合物中的多种产物,并获得比以前报道的文献更广泛的反应条件。
2010 年,Buchwald 和 Jensen 等人描述了一种基于自优化自动化流的微反应器系统,该系统采用在线 HPLC 监测反馈控制和由“黑匣子”算法指导的优化算法。该系统被证明可以最大限度地提高 Knoevenagel 缩合 [206]、苯甲醇和苯甲醛氧化 [206] 以及 Heck 反应 [230] 的产率。该小组使用了与在线红外监测相结合的类似策略来优化 Paal-Knorr 反应 [231]。 2016年,基于序贯自适应响应面法(ARSM)结合优化实验设计,他们开发了一种自动反馈液滴流微流控系统,涉及离散变量,即前催化剂和配体,以及连续变量、温度、时间、和加载,探索和优化 Pd 催化的 Suzuki-Miyaura 交叉偶联反应 [232]。
Cronin 报道了一种集成的自优化合成流平台,该平台结合了用于监测和控制有机反应的实时在线台式 NMR,并采用改进的 Nelder-Mead 算法 [233] 进行自优化 [234]。 Felpin 的团队还使用改进的 Nelder-Mead 优化算法在具有各种目标函数的四维空间中执行 Heck-Matsuda 反应优化 [235],以及天然产物 Carpanone 的自优化合成 [236]。最近,Felpin 的小组描述了一种自动连续流装置,使用自制的光化学反应器,通过光硫醇烯反应对金鸡纳生物碱与各种有机硫醇进行自我优化和功能化[237]。利用他们之前开发的 Nelder-Mead 方法的修改版本,在 3 维空间中对反应进行了优化,即停留时间、停留时间的当量以及基本负载。
2016 年,Bourne 的团队记录了一个自动化连续反应器,集成了在线 MS,用于反应监测和产物定量(图 8.6)[238]。由于其快速定量(< 1 分钟分析持续时间),在线 MS(使用线性相对响应值通过 HPLC 进行校准)可以提供化合物和中间体的实时定性鉴定或相对成分分析。他们很快描述了一个类似的自动化反应器和自适应反馈控制,用于优化合成奥西替尼(一种不可逆表皮生长因子受体激酶抑制剂)的最后一步[239]。自优化流动反应器将在线 HPLC 分析与反馈 SNOBBFIT 算法相结合,可快速达到最佳条件。在他们的研究中,SNOBFIT 在每个优化周期中生成的条件按温度升高进行排序,以最大限度地减少加热和冷却所需的时间。一旦反应器达到所需温度,所有泵均设置为每分钟 0.02 mL,并设置正确的流速。使用产物和内标之间的 HPLC 峰的比率计算实时收率。系统将不断循环,直到找到最佳条件(图 8.7)。

图 8.6 带反馈控制和优化算法的自动流动反应器

图8.7 自优化算法工作流程
Lapkin 和同事开发了一种基于模型的实验设计 (MBDoE) 和自优化方法,用于流动中的 Csp3-H 激活(图 8.8)[240]。利用在线 UV 和在线 GC 模块进行实时分析。
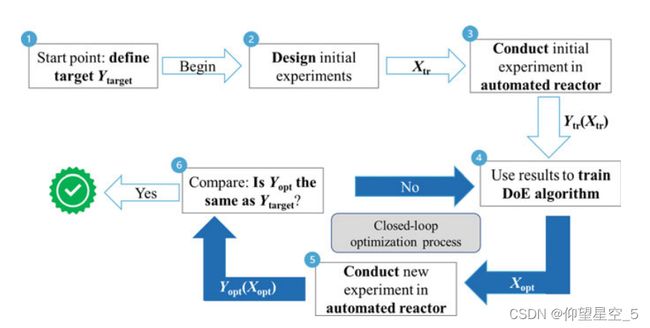
图 8.8 MBDoE 激活 C(sp3)–H 的闭环优化过程
Zare 的团队开发了一种模型,即深度反应优化器 (DRO),通过利用深度强化学习和化学领域知识来指导化学反应优化中的交互式决策过程 [241]。使用MS的输出作为优化目标,DRO可以迭代地与化学反应交互以获得当前的反应条件和收率,然后确定要尝试的新实验条件。
2018 年,Bédard 等人开发了一种完全集成的多功能系统,用于条件优化和基质范围评估 [242]。这种方便的可重构系统成功演示了许多反应,包括 Buchwald-Hartwig 胺化、Horner-Wadsworth-Emmons 烯化、还原胺化、Suzuki-Miyaura 偶联、亲核芳香取代 (SNAr) 以及可见光光氧化还原反应。该平台还用于执行涉及有限稳定乙烯酮中间体的两步过程。
基于之前的研究,Jensen 等人于 2018 年实施并验证了混合整数非线性程序 (MINLP) 算法,通过同时调节催化剂类型和连续变量等离散变量来优化催化剂周转数 (TON) 和产品收率,包括温度、停留时间和催化剂负载量[243]。由于能够在存在副反应的情况下确定最佳条件,该算法随后被应用于自动化微流体反应平台中Suzuki–Miyaura交叉偶联反应的实时优化,该平台包括液体处理器、振荡流反应器、和在线 LC/MS。
Pentelute 和同事通过在线 UV-vis 分析对从 35,427 个高度可重复的脱保护步骤收集的数据应用深度学习 (DL),以预测肽合成并最大限度地减少易于聚集的肽的聚集 [244]。这种深度学习方法还能够根据看不见的数据预测序列的合成结果,这表明它能够预测和优化自动流程合成平台中的肽合成。
最近,Collins等人设计了一种自动化多步化学合成仪AutoSyn,包括固定配置的基于流程的合成平台、试剂输送系统和过程分析技术,以及用于自动端到端过程操作的集成软件控制系统和监测(图8.9)[245]。该合成器能够在几个小时内组装出毫克到克级的几乎类似药物的小分子。

图 8.9 AutoSyn 示意图
Cronin 的小组提出了另一个闭环反应寻求神经网络驱动的自主机器人平台,用于发现新反应(图 8.10)[188]。Cronin 和同事设计的名为 Reactify 的卷积神经网络能够通过 NMR 光谱为“看不见的”反应混合物分配反应性值,并与一个闭环系统相结合,用于探索实验空间的反应性。通过该系统识别了一系列化学反应和产物,包括几种新反应以及新的反应模式。

图8.10 神经网络驱动的自主平台新反应发现
合成化学家通常使用一小组相似的起始化合物来探索反应。然后,将最适合这些模型底物的优化条件应用于许多其他通常不同的化合物,即使替代条件可能会改善结果。最近,Grzybowski 和 Burke 等人报道了 ML 和自动合成的迭代方案,以探索一般反应条件 [246]。这个简单的闭环工作流程利用数据引导的矩阵下选择来使大型搜索空间易于处理,同时保持整体的有效性,利用不确定性最小化机器学习来有效地驱动预测优化,并采用机器人实验来提高吞吐量、精度、以及按需递归生成的数据集的可重复性,为具有巨大搜索空间的多维化学优化问题提供了实用的解决方案。该工作流程成功确定了(杂)芳基 Suzuki-Miyaura 交叉偶联反应的一般反应条件。
仍然需要人工输入来填写关键程序细节并手动优化 CASP 生成的近似反应条件。 2022 年,Jensen 的团队报告了一种算法引导的多步反应优化方案,以确定 CASP 生成的近似合成配方的最佳工艺条件 [247]。他们使用 ASKCOS(一种基于计算机辅助合成的多目标贝叶斯优化算法的开源 CASP 软件 [58,176,248–250])来迭代生成目标分子的合成路线和反应条件的建议。经过人工评估合成可行性后,自动合成平台选择并完成了高级合成途径。由于可用数据有限,仍然需要人工干预。
当前的机器学习实现依赖于大量数据集的利用以及黑盒算法和回顾性基准统计。此外,考虑到有效处理非常规任务需要专业化学知识,大多数算法在有机化学实践中的适用性存在问题。 2020 年,伯纳德斯和罗德里格斯等人开发了 LabMate.ML,这是一个计算框架,通过利用无偏见的随机实验选择进行初始化和自适应机器学习来导航未知的反应空间。 LabMate.ML 选择小数据进行初始化,例如 5-10 个随机反应(搜索空间的 0.03-0.04%)(图 8.11)。然后进行反应,并将收集到的数据视为初始知识库。随后的反应在计算机建议的条件下进行,基于主动学习启发法,并通过液相色谱-质谱/紫外-可见光(LC-MS/UV-vis)进行评估。所需 m/z 或 UV 吸收峰的曲线下面积 (AUC) 值用作反应结果(例如总产物量或产率)的代理,被视为机器学习中的客观目标值。反应条件用作 LabMate.ML 的特征,以在优化器中进行训练。 LabMate.ML 使用 RF 生成模型,使可用数据合理化并建议新的条件集。该软件是自主的,并且在每次迭代后都可以重新训练。 LabMate.ML 只需 5-10 分钟即可在个人计算机上运行完整的重新训练周期和预测。

图 8.11 LabMate.ML 反应优化的工作流程
5. 合成自动化、机器人化学家和云实验室
自动化提供了将有机合成从劳动密集型工作转移到机器驱动过程的途径[251]。自动化合成的第一个概念可以追溯到 20 世纪 60 年代,当时 Merrifield 和 Stewart 报道了固相肽合成的自动化系统 [252-255]。自动化系统获得的肽在产量、纯度和生物活性方面与手动合成的肽相当,同时时间和材料损失显着减少。利用类似的策略,可以通过自动化程序合成 DNA [256, 257]、RNA [258] 片段以及多糖 [259, 260]。
肽和寡核苷酸自动合成仪中的合成方案对于每个分子来说基本上是相同的。相比之下,通用的自动化有机合成仪器需要将合成路线调整为整体的、相互依赖的和多步骤的过程,这些过程对于每个分子来说大多是独特的[245]。鉴于化学研究中的自动化很少见,而且商用系统通常是为特定目的而设计的,并且仅对重复性工作有效,1978 年,Legrand 和 Foucard 为合成化学家开发了一套自动化套件 [261]。执行器可以执行各种操作,例如加热、冷却和试剂添加。传感器可以测量各种参数,包括温度、压力、体积、重量、pH 值和氧化还原电位。
Ley 的团队设计了一种方便高效的原型,用于在连续流动过程和批量模式下蒸发、浓缩和切换溶剂 [262]。该系统已成功应用于伸缩过程。 2013 年,艾伯维公司的研究人员开发了一种高效的化合物合成系统,具有集成组件和自动样品处理模块 [263]。该系统仅用了三天时间就准备好了包含 48 个成员的 10 mg 化合物库,包括合成、纯化、验证和样品分配。 Tu 等人很快开发了一个基于 SWAVE 平台和内部开发的机器人技术的全自动合成纯化站 [264]。选择两个广泛使用的反应,即 Sonogashira 偶联和 Buchwald-Hartwig 胺化,作为集成系统有效性的证明。 最近,Ahmed 的团队开发了一种机器人辅助声流体末端执行器 (RAEE) 系统,由机械臂和声流体末端执行器组成 [265]。 RAEE 系统通过流体泵送、颗粒和斑马鱼胚胎捕获以及孔板中复杂粘性液体的移动混合进行了演示。
连续流制造现已在活性药物成分(API)以及其他精细化学品的合成中被广泛接受[266-269],无论在实验室还是制药行业,都是一个高效且有吸引力的技术平台[269-273] 。由于其固有的优势,例如反应时间、温度和成分的精确控制,连续流化学是计算机控制合成的理想技术,迄今为止已经报道了大量自动化连续流平台的例子[21 、26、216、217、269、272、274、275]。例如,2010年,Machida等人开发了一种溶液相自动合成仪ChemKonzert,用于制备多种有机化合物[276]。系统中集成了离心分离器,以实现高效的液液萃取。进行了几个例子来证明这种自动合成器的性能(方案8.5),包括通过36步路线合成浆果赤霉素III[277],紫杉醇的关键中间体,9元掩蔽烯二炔[278],加纳醛[279],广泛使用的多步合成中间体。
图8.5 ChemKonzert应用实例
Pentelute 的实验室于 2017 年报告了一种基于流程的自动化多肽合成系统 [280]。该方法仅需 40 秒即可完成氨基酸残基的全合成。作者声称,通过这种方法,每年可以满负荷生产数以万计的 30 聚体肽。同样,辉瑞公司的研究人员开发了一个基于流程的自动化合成平台[103]。该系统将快速纳摩尔级反应筛选和微摩尔级合成集成到一个模块化单元中,通过探索高温下纳米级铃木-宫浦耦合中一系列不同的反应变量进行了验证,并且能够执行超过每 24 小时 1500 次实验。最近,Li 和同事发明了一种名为 Tiny Tides 的全自动快速流动设备,与之前报道的方法相比,该设备可以将偶联时间减少多达 22 倍,从而生产按需定制的反义二酰胺磷酸吗啉低聚物 (PMO) [281]。这种高效合成器还可以单次合成细胞穿透肽-缀合肽-核酸(PPNA)[282],相比之下,肽-核酸(PNA)单体之间每个酰胺键的形成只需要10秒对于微波肽合成仪,每个酰胺键 10 分钟;或每个耦合周期 32 分钟,就像 DNA 合成器 Expedite 8909 一样。他们还使用 Tiny Tides 的合成数据来训练有效的 ML 模型,以指导高效的 PNA 序列设计 [283]。
2020年,Dömling团队使用基于正压的小批量非接触式点胶技术I-DOT,通过Ugi-3组分反应以全自动方式合成了1000多种不同的亚氨基吡咯烷-2-羧酸衍生物。纳米级[284]。 1152次反应,贵重试剂和溶剂的总化学消耗量小于0.6毫克,而在毫摩尔规模进行相同数量的反应时,大约需要400克试剂和12升溶剂。
Williams 和 Kappe 等人最近设计了一个完全集成的多步反应和实时分析平台 [285],该平台用于以完全受控和稳健的方式合成 API 美沙拉嗪(5-氨基水杨酸,5-ASA),超过三个合成步骤(硝化、氢化和水解)和三相分离(一次气/液分离,两次液/液分离),吞吐量为 1.6 克/小时(方案 8.6)。先进的数据处理技术增强了四种实时分析工具的使用,包括在线 NMR、在线 UV/vis、在线 IR 和在线 UHPLC。
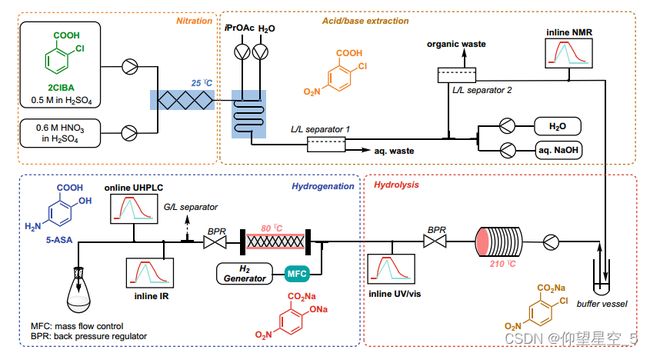
方案 8.6 5-ASA 的 PAT 工作流程
一般的流程方法基于线性策略(图8.12a),这总是导致效率低下。 2021年,Kim的团队开发了一种平行流合成器,能够进行多重合成和化合物库优化(图8.12b)[286]。与线性流方法相比,这种并行流合成器要快得多。合成器可以在流线堵塞时不间断地执行其功能。它可以通过采用不同的进料速率、浓度和温度等反应参数来独立控制配置的毛细管。利用该平台,Kim等人在不到一小时的时间内从96个不同条件中筛选并获得了芳香族取代反应(C-C、C-N和C-S键)和偶氮偶联反应的最佳条件。鉴于大多数报道的自动化多步合成过程依赖于迭代或线性程序,并且需要在多功能性和设备方面进行折衷,Gilmore 和同事详细介绍了一种基于围绕中央核心排列的多个连续流模块的自动径向合成器(图 8.12) c) [287]。该平台允许远程控制,并且可以稳定且可重复地提供线性和收敛合成,而无需手动重新配置。此外,该自动化仪器还集成了红外和核磁共振光谱仪,用于在线监测,从而实现反应后分析和反馈。
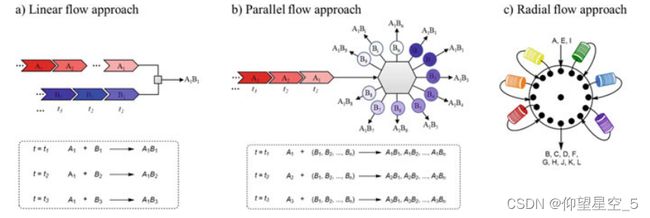
图 8.12 不同类型的流程。一般流程方法。 b 平行流方法。 c 径向流法
由于固体常常导致反应器通道堵塞,因此在研究规模的流动反应器中处理固体仍然很困难。尽管已报道使用除酸有机碱在质子化时生成低熔点至中等熔点的离子液体(IL)作为防止堵塞的解决方案[288],但这些添加剂可能会使反应混合物复杂化或引起副反应。 2022 年,Jensen 等人开发了一种连续搅拌釜反应器 (CSTR) 流动平台,可以在流动化学转化过程中处理浆料和固体 [289]。他们还采用 MINLP 进行单一自动优化,并采用贝叶斯优化算法进行自动多目标优化例程。这种用于通过固体/浆料进料和处理进行反应自我优化的全自动流程平台将加速识别制造工艺开发的反应条件优化。
目标产物的分离和纯化是有机合成中必不可少的步骤。流动化学的理想过程是将反应物输入流动系统,并从过程中输出纯产物(方案 8.7a)。 George 等人展示了一个这样的例子,他们使用超临界 CO2 流动系统连续合成青蒿素 [290]。然而,事实是,在进行下一步之前,必须中断许多多步反应流系统进行后处理和萃取,以去除副产物(方案 8.7b)。随着在线固相萃取[291]、气液和液液分离[292]技术的发展,这些后处理中的大多数都被纳入单个连续过程中(方案8.7c)。然而,反应后仍需要离线纯化,通常是柱色谱法,以获得纯的最终产物。 Baranczak 和同事开发了一个全自动集成平台,用于加速小分子库的合成-纯化-测试(方案 8.7d)[293]。该平台能够基于广泛的化学转化进行批量支持的化合物合成,并以各种生化分析格式进行测试。利用 HPLC-MS-CAD(带电气溶胶检测器)集成来分离和测定所需产物的纯化级分的浓度。通过文库合成产生的化合物足以在二次测定中进行进一步评估,从而大大缩短药物化学发现周期。最近,Lee 和 Vilela 等人报道了一种在线色谱纯化自动流动合成平台,通过连续分离最终产物来加速自动流动合成(方案 8.7d)[294]。在该系统中,三个不同的反应成功应用于在线正相或反相快速色谱法,连续分离产品的纯度为 97-99%。

图式8.7 流动化学中产品分离纯化的多样性
Burke 采用类似于迭代肽偶联的统一且完全自动化的过程,通过 Suzuki-Miyaura Csp2-Csp2 键形成获得 14 种不同类别的小分子(方案 8.8)[295]。这种类似乐高的合成方法使用 N-甲基亚氨基二乙酸 (MIDA) 作为构件,组装到起始材料的硼酸酯基团中,以降低硼酸的反应性,从而防止发生不良反应。此外,当使用四氢呋喃 (THF) 作为洗脱剂时,MIDA 硼酸盐将选择性地从硅胶中洗脱[296, 297],从而可以在反应后通过“捕获和释放”方案轻松纯化。由于已经有超过 5000 种兼容的构建模块可供商业使用,因此该合成机器可以合成大量的小分子。然而,这种迭代组装策略与立体特异性 Csp3-Csp2 或 Csp3-Csp3 成键反应不相容,因为 MIDA 硼酸酯对 Csp3-C 成键条件敏感,需要水性碱性条件或亲核试剂来与MIDA 硼酸盐。最近,Burke 的团队开发了一种新型四甲基 N-甲基亚氨基二乙酸 (TIDA) 硼酸酯,该硼酸酯在 Csp3–Csp2 和 Csp3–Csp3 键形成的条件下稳定。最近,Burke 的团队开发了一种新型四甲基 N-甲基亚氨基二乙酸(蒂达)对 Csp3-Csp2 和 Csp3-Csp3 键形成条件稳定的硼酸盐 [298]。新方法允许通过自动合成来组装 Csp3 硼酸酯构件,包括通过自动立体特异性 Csp3-C 键形成来合成天然产物。小分子迭代合成的方法已进行了综述[20, 299]。

图式8.8 C-C耦合的自动迭代合成策略
微流控形式是 HTE 技术的一种小型化流动类型。 Jensen 和同事开发了一系列液滴微流体自动化平台 [300–305]。例如,2017年,他们报道了一种基于液滴的自动化分段流动化学平台,能够进行高效反应筛选、小规模产品分离以及在100 μg规模下量化多种药物化学物质[300]。 2019年,Jensen和Buchwald等人利用自动化微流控平台在可溶性有机碱存在下优化Pd催化的C-N偶联条件,涵盖各种N-亲核试剂和芳基三氟甲磺酸酯[301]。基于优化算法,他们利用了许多配体和碱基组合来进行这种转化。他们还开发了一种自动化微流体单液滴筛选和分析平台,该平台支持自动化 HTE,以加速新有机电过程的发现 [302]。 2020年,Kennedy和Stephenson等人开发了一种液滴微流控平台,该平台能够实现基于流的高通量反应发现,以建立药学相关化合物库[306]。该平台允许在皮摩尔规模(即 10-12 mol)上进行反应,并通过在线电喷雾电离质谱 (ESI-MS) 收集高通量数据,以每秒 0.3 个样品的采集速率进行快速分析。最近,Jensen 团队与 Pidko 等人合作,通过自动化微流体平台报道了敏感 β-氨基酮底物的催化不对称氢化反应 [305]。
在制药行业中,通常会利用各种来源的分子片段来组装 API,然后在不同的工厂中形成最终的药品,这导致人们对 API 和药品的连续生产越来越感兴趣,并且此类材料储存和运输的安全问题[307]。此外,批量生产的局限性,例如质量控制的变化和供应链中断[308, 309],通常会导致药品短缺。为了解决这些问题,2016 年,Adamo 及其同事开发了一个连续制造平台,将合成和最终药物产品配方结合到一个高度紧凑的单元中 [310]。冰箱大小的连续端到端合成系统实现了高效的传热和传质,以及过程强化和自动化。该系统每天可以生产数百至数千种符合美国药典标准的口服或外用液体剂量的高品质 API。
2021 年,Bode 的小组报告了一种集成系统,用于在最少的人类参与下自动基于胶囊合成饱和 N-杂环和还原胺化 [311]。所报告的控制台结合了试剂设计、硬件工程和简单的操作系统,可以使用预包装的胶囊执行复杂的有机反应,并通过以全自动方式执行多步骤序列来提供药物样分子。
虚拟筛选是一种用于药物发现的计算技术,用于自动搜索和评估异常大的小分子库,以识别那些最有可能与药物靶点结合的结构[312]。然而,由于虚拟筛选活动中误报率较高,必须合成潜在命中进行验证,这在手动完成时既费时又费力。 2022年,Bode等人报道了一种迭代自动合成控制台,能够从超过7×1012个成员的虚拟库中快速组装分子,其中包含具有类药物理化特性的新型支架[313]。与手动工作流程相比,这种方法提供了十倍的效率提高,这表明这种自动合成控制台可以促进从大型虚拟库中快速且可靠地准备有吸引力的虚拟筛选命中。
Cronin 等人提供了另一种协议来缓解上述问题[314]。他们报告了一种方法,可以在一系列互连的塑料模块中定制合成目标化合物的蓝图,这些模块可以通过使用三维(3D)打印技术来制造。这些设备是使用化学计算机自动化设计(ChemCAD)方法设计和构建的,该方法能够将传统的小规模合成程序转换为独立于平台的数字代码。 γ-氨基丁酸受体激动剂和(±)-巴氯芬的成功演示展示了这种方法在专业设施之外本地生产药物的可能性。 3D 打印反应件中的这种数字化合成方法也应用于其他各种示例中 [315, 316]。最近,Cronin 等人报道了一个便携式手提箱大小的平台,其中包含通用化学合成的合成和纯化所需的所有模块[317]。该系统使用化学标记语言 (χDL) 与数字反应器生成器耦合,通过 3D 打印和基于文本文献合成的可执行协议来生产反应器。这个迷人的平台为未来的自动化合成平台设计提供了蓝图。
尽管综合自动化和人工智能得到了发展,但人工较少、可远程控制和监控的全自动化智能平台或实验室仍然存在需求。在COVID-19大流行初期,全球大多数实验室因公共卫生原因而暂时关闭,这极大地影响了科学研究的进程,特别是有机化学等实验性学科。因此,建设“云化学实验室”,即允许化学家远程进行化学实验,是一个非常有吸引力的想法。事实上,一些研究小组已经探索了建立“云实验室”的可能性。 Poliakoff 的团队展示了一个概念验证,即研究人员可以远程操作自优化系统 [318]。 2016 年,Ley 的团队构建了一个基于互联网的模块化软件系统 LeyLab,使化学家能够通过互联网使用世界各地的任何设备监视和控制化学反应 [319]。它促进了合成过程的自动化,并且能够自主地自我优化反应参数,以找到满足可定制的多组分优化功能的最佳条件。无论计算机知识背景如何,研究人员都可以享受该系统的好处。 LeyLab 的实用性通过应用反应自动化来最大化固定体积催化剂的输出以及针对各种目标函数优化三维多相催化反应和五维阿佩尔反应来证明。
2018 年,Aspuru-Guzik 和同事描述了一种名为 ChemOS 的便携式模块化软件框架,用于操作具有集成机器人、传感器和人工智能的闭环系统 [320]。这个单一软件框架适用于许多不同的任务,包括学习染料的颜色空间和自动校准高性能液相色谱分析。
Cronin 的团队很快开发了一种架构和抽象,Chemputer,它将报告的方法指令映射到适合使用传统实验室规模技术自动化的各个步骤(方案 8.9)[321]。在他们的研究中,自动化单元是在机器人平台上实现的,该平台运行编译的化学语言来控制分子的合成。即使用户缺乏编程知识,他们也可以毫不费力地编写必要的语言。作为对这一概念的验证,Chemputer 在没有人工干预的情况下组装了三种高质量的药物化合物,其产量和纯度比手动实现的要好。 Cronin 的团队还使用自然语言处理 (NLP) 算法,提出了一种系统,可将化学文献中的合成过程数字化为可在自动合成平台上执行的化学代码 (χDL) [37, 322–324]。
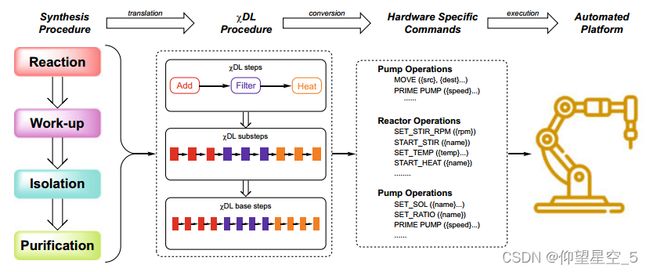
图式8.9 化学计算机示意图
2020年,朱的团队报告了一种智能机器人系统,即材料加速操作系统(MAOS),用于材料合成和科学发现[325]。人类化学家可以通过4G/5G网络通过VR与机器人交互来监测和控制系统。对于训练后的实验,MAOS 可以通过全自动程序和人工智能保证的质量控制来完成实验任务。
尽管人们从 20 世纪 80 年代就开始探索化学实验室机器人的可能性[326-328],但第一个人工智能集成移动机器人是由 Cooper 团队于 2020 年开发的[329]。这位贝叶斯搜索算法驱动的机器人化学家可以在八天内自主操作,在十个变量的实验空间内进行 688 种反应。然而,由于缺乏计算大脑,机器人搜索既无法捕获现有的化学知识,也无法生成或评估科学假设。
Jiang 的团队最近记录了一位全能的人工智能化学家,它能够执行化学研究所需的所有基本步骤 [330]。基于服务平台,AI-Chemist可以自动从云端数据库中读取文献,并据此提出实验方案。它可以通过控制移动机器人来执行实验过程,同时通过机器学习和贝叶斯优化由计算大脑分析实验数据。凭借“科学思维”,这位人工智能化学家还可以为下一次迭代提出新的假设,这展示了其像训练有素的人类化学家一样进行高水平化学研究的能力。
6. 展望
想象一下,您在周一早上醒来,坐在桌旁享用咖啡。突然您注意到最近在线的一份科学出版物称最近刚刚分离和表征了一组新的生物活性天然产物,其全合成仍在研究中。您兴奋地拿起手机,向实验室机器人的服务器发送一条命令行,然后继续吃早餐。然后你开车去上班。当你进入实验室时,那组新发现的天然产物已经放在通风柜上的小瓶中,上面贴着它们的名称、结构、纯度和重量的标签——你的实验室机器人在收到信息时已经自主合成了相应的化合物。早上你的命令。你冲到办公室,检查桌上的报告。它涉及这些天然化合物的合成细节和特征,以及全合成过程中发现的两个新反应,包括所有优化细节、底物范围扩展和机理研究。仔细检查数据后,您开始准备并提交有关这些天然产物的首次全合成和两个新反应的发现的手稿。经过两周的同行评审后,您的手稿将被接受并在高影响力期刊上在线发表。只需单击一下手指,所有合成的事情都将完成。对于合成化学家来说,这是一个多么令人兴奋的梦想。但让我们回到现实。尽管有很多令人印象深刻的例子展示了机器学习和合成自动化在当今和未来合成化学中的力量和无限可能性,但仍有一些问题和担忧需要解决,如下所述。
6.1 数据可用性和质量。
作为机器学习的重要组成部分,数据可以通过 SciFindern 或 Reaxys 等在线数据库访问。然而,现有数据集存在一些局限性:
(i) 信息缺失。反应细节总是被省略。例如,许多出版物没有报告温度、压力、溶剂消耗等条件细节,这些可能是转化成功的关键变量。此外,化学家通常非常关注报告产率,这意味着立体选择性或区域选择性经常在出版物中被丢弃,即使这些数据有助于训练选择性预测模型。
(ii) 数据偏差 [72, 73]。研究人员总是更喜欢展示他们的成功结果,而将负面结果远离公众,尽管这些失败对于设计未知化合物的可靠合成路线至关重要[17]。这导致在使用此类数据集之前需要进行广泛的数据清理和管理[331]。
(iii) 数据质量。尽管此类在线数据库中的数据是从同行评审的出版物中提取的,但无法保证数据的可靠性和可重复性[332]。与生物学家不同,化学家很少重复其他研究小组发表的实验来验证实验结果。
因此,我们宁愿相信高影响力期刊上发表的结果。然而,在如此高影响力的期刊中,撤稿仍然发生。此外,研究论文中还会出现拼写错误等微小错误。例如“CuCl”和“CuCl2”,“0.2 mmol/L”和“0.2 mol/mL”等。有时这样的错误可以忽略不计,但有时这个微小的错误会指向相反的一面。 (iv) 大多数在线反应数据库都是商业性的,这给用户访问设置了障碍。
获取机器学习数据集的另一种方法是由自动机器而不是研究生进行内部实验。借助 HTE、流动化学和相应的自动化技术,研究人员可以轻松快速地获得大量数据集。许多案例已经证明了这些综合自动化的生产力。由于合成要求不同,每个实验室的平台和程序可能不同。问题是,从不同实验室获得的数据如何相互通信?或者更具体一点:我们如何处理我们的实验记录,使其变得详细、可追溯、可共享?一种解决方案是使用电子实验室笔记本(ELN),它在工业界广泛使用,但在学术界仍然很少见[17, 333]。但随之而来的几个问题是:(i)每个群体对 ELN 系统都有自己的偏好;有些甚至使用自制的 ELN。不同的 ELN 系统中的数据如何传输到另一个系统? (ii) ELN 系统大多是商业化的。对于那些负担不起的实验室那么他们如何与他人分享他们的研究数据呢? (iii) 当研究人员被要求共享尽可能多的原始数据时,如何平衡共享内容和研究隐私?
最近,Kearnes 等人提出了构建和共享有机反应数据的范例[334]。此类开放访问数据库,包括集中式数据存储库,即开放反应数据库(ORD),可在 GitHub 上公开获取。出版物中的支持信息 (SI) 将被标准化,并且任何人都可以在 ORD 模式下阅读。此外,重复性所需的程序细节的省略将更加明显,而反应之间的比较将更容易。 ORD 格式和其他新兴数据标准 [98, 335] 以及 ELN 格式之间的转换工具正在发布。 ORD 是化学界反应数据存储库的一个辉煌的开始和尝试。鉴于超出要求或同行评审出版物的数据共享在化学研究中并不常见,像 ORD 这样的数据存储和共享平台仍然面临社会和文化障碍。另一方面,化学家应该克服不愿意分享低产率或选择性的不满意结果的心理,无论自然反应性、程序错误或人为错误如何。
6.2 分子和反应的表示。
描述符对于机器学习至关重要。描述符过多或不足是否会限制预测模型的预测能力[30]。正确的描述符可以带来预测模型的优异性能[336],例如狄尔斯-阿尔德反应的主要区域、位点和非对映选择性结果预测[337]。因此,应该谨慎选择合适的方式来表示分子。如上所述,建立机器学习模型有多种表示分子和反应的方法。虽然DFT和MM计算可以提供大量的描述符,但每个分子的量子计算将花费大量的时间和计算机资源预算。此外,由于每个分子支架的物理参数各不相同,因此创建全面的描述符集仍然是一个挑战[30, 162]。另一方面,目前的描述符在描述一些分子支架(特别是有机金属配合物)以及分子的立体电子特性和 3D 构象方面遇到了障碍 [338]。
将分子或反应翻译成自然语言(如句子)是表示分子和反应的另一种选择。事实上,使用自然语言处理(NLP)的 Bert RXN 和 AutoGluon Text Prediction 等算法已经被用于反应预测 [339-341]。然而,分子结构和反应性之间的关系很难通过自然语言处理来反映,这限制了此类方法在反应预测中的应用。因此,开发新的描述符来准确、简洁地表征分子和反应是未来研究的重要组成部分。
6.3 预测的适用性和可靠性。
尽管当前的反应预测模型(包括逆合成计划)已经显示出了出版物所声称的令人难以置信的预测能力,但我们想问的是,如果将此类预测模型应用于更大的化学空间(适用性),化学家相信的可能性有多大?即使模型的评估指标如出版物所述令人印象深刻(可靠性),人工智能仍能做出预测结果?
对于前者,根据“没有免费的午餐定理”[342],不存在解决所有问题的单一最优算法。因此,关于预测适用性的声明至关重要。例如,报告反应结果预测模型的团队最好通过外部验证(当然是通过合成自动技术和 HTE)找出模型的适用空间,这可以使他们的模型更加实用而不是理论,而不是简单地声称他们已经开发了一个预测模型但没有任何证据。化学家应该认识到,发表论文并不是科学的目的;而是追求真理。
最后一个问题有点复杂。预测反应结果(例如产量、选择性和可能的产物)的模型很容易得到验证,因为单个反应就足以说明问题。对于逆合成规划来说,问题要复杂得多,因为只有在最终产品成功合成或中间步骤失败的情况下,预测的合成路线才会被证明是可行或不切实际的,这是一个消耗资源的过程(时间、人力、金钱)。此外,如果逆合成规划模型提出了一种超出化学家意识的新策略,而不是已知的转变,那么化学家处理这种合成路线的可能性有多大?提供可靠且广泛接受的预测模型应该是有机智能社区的目标。人工智能驱动的全合成的成功例子越多,化学家选择相信预测模型的机会就越大。
6.4 化学家的计算机背景。
有机合成自动化、智能化的趋势是不容置疑的。当这些年有机化学跨机器学习的出版物不断涌现时,有人可能会嗅到有机界的焦虑和恐惧,因为大多数有机化学专家在编程和机器学习方面也是新手。上述大多数人可能会关心两个问题:(i)传统化学家是否会被新兴的人工智能和机器人取代? (ii) 缺乏相应背景的化学家如何开始涉足机器学习领域?对于第一个问题,答案是否定的。事实上,传统合成程序中的人类行为将逐渐由自动化机器和人工智能来代替,因为与手动工作相比,合成自动化和人工智能可以实现更高效、更可靠的结果。然而,自动化机器和人工智能的普及肯定会将化学家从日常的手动任务中解放出来,使研究人员能够更多地关注研究的创造性方面并改善他们的工作与生活平衡。此外,通过使用合成自动装置(或机器人),化学家可以在更安全的情况下进行反应,特别是当涉及有毒或爆炸性化学品时。此外,自动化和人工智能技术的兴起使研究人员能够继续他们的调查,而不会因 COVID-19 期间实验室关闭等情况而中断。人类化学家是不可替代的。相反,我们将在未来的合成化学领域扮演另一个角色。我们应该从“如何”转变为“什么”和“为什么”:从思考“如何合成这个分子”转变为“我们应该合成什么,为什么要合成它”。
没有相应背景的化学家在第一次开始机器学习时可能会感到压力。实际上,对于没有编程背景的化学家来说,有一些用户友好的工具可以在他们的研究中使用机器学习。例如,AutoGluon [160] 使 ML 初学者和专家能够通过几行 Python 代码快速构建高度准确的 ML 模型。通过自动超参数调整、模型选择/集成、架构搜索和数据处理,这个开源框架允许化学家在他们的研究中轻松使用机器学习。使用 AutoGluon 构建反应预测模型的示例已被记录在案 [161a](见上文)。对于逆合成规划者来说,开源(例如 AiZynthFinder、ASKCOS、IBM RXN for Chemistry)和商业(例如 Chemical.AI、ICSYNTH、Reaxys、SciFindern、Spaya、SYNTHIA)平台均可使用。此外,免费的开源数据分析平台 KNIME 可以帮助没有专家背景的化学家分析数据以及执行机器学习代码。我们相信,随着这些适合初学者的工具的发展和普及,编程背景将不再成为化学家和机器学习之间的障碍。
6.5 整体自动化和便利的机器人。
通常,自动化合成平台包含以下模块:试剂存储、反应准备模块、反应器、分析和纯化系统、化合物管理系统、监控单元和控制台。为了减少执行自动合成时的人为干预,这些模块应该无缝集成。同时,考虑到大多数研究小组没有足够的空间容纳大型机器,这些高度集成的自动化设备应该具有合理的尺寸,理想情况下小到通风柜,甚至手提箱大小[317]。此外,由于每个实验室对实验室自动化的需求各不相同,并且并非所有化学家都具备自动化知识,因此自动化系统应该高度且易于定制。一个好的自动化平台不仅应该满足用户的需求,还应该在使用过程中具有适应性。此外,无论硬件还是软件,理想的自动化平台都应该使用方便、简单,并且可以远程控制。考虑到大多数合成化学家没有自动化或编程背景,因此需要一种基于化学家用户习惯的通用化学编程语言。最后但并非最不重要的一点是,自动化设备(包括硬件和软件)的设计、生产和维护需要大量的时间和金钱,这对于大多数实验室来说是无法承受的。机器可以日夜运行,没有任何抱怨。但如果在使用实验室自动化方面的支出远多于雇用人类操作员,那么大多数实验室宁愿为人力而不是机器人付费。因此,降低自动化平台的配置成本,将有助于自动化平台的普及,特别是在基层的普及,更好地促进化工自动化的发展。用于液体处理的可定制且用户友好的实验室机器人范例已由 Opentrons 商业化。正如他们声称的那样,他们的商业实验室机器人和工作站(OT-1 和 OT-2)是开源的,易于定制,价格低至 10,000 美元。
6.6 使用合成自动化和机器学习的成本。
将新手训练成机器学习/自动化专家显然需要时间。研究生花费数年时间成为熟练的化学家,没有人抱怨这花费的时间。为什么他们如此关心转向机器学习和综合自动化所需的时间?随着用户友好的自动合成平台的普及和可行机器学习工具(例如 AutoGluon,见上文),我们认为培训初学者使用合成自动工具和机器学习并不耗时。相反,合成自动技术和机器学习的使用肯定会加速调查过程,并获得明显更可靠和可重复的结果。
支出是另一个可以避免的问题,因为几乎每个实验室的预算都是有限的。如上所述,部署自动化合成平台或实验室机器人非常昂贵。过去,化学家将大部分预算花在购买化学品、玻璃器皿、反应器以及其他分析和实验设备(例如 GC-MS、旋转蒸发器等)上。由于合成平台主要由多个模块集成,并且合成平台上的反应规模比传统合成小,化学家可以调整化学品和设备的成本来建立合成平台。同时,我们希望像OT-2这样更经济的自动化合成模块能够进入市场,这可以减轻那些想在自动化合成大道上驰骋的化学家的经济负担。
另一方面,机器学习的主要成本应该是计算资源。虽然有时一台 PC 足以训练预测模型,但精确的模型总是需要更好的计算能力,尤其是在涉及量子计算时。一个解决方案是使用云服务器。云服务器是其他人(超云提供商)的计算机。超云提供商可以提供更低的总拥有成本,同时提供从可扩展性到安全性的卓越功能。云服务器可以帮助化学家获得最新的图形处理单元(GPU)甚至张量处理单元(TPU),而这些是化学家自己几乎无法负担或维护的。 Amazon Web Services (AWS)、Google Cloud Platform (GCP)、IBM Cloud、Microsoft Azure、阿里云和腾讯云是几个著名的 ML 云平台。这些云服务器的使用将为那些预算有限但渴望推动机器学习铺路的化学家释放压力。在“网络化学时代”,有机合成是毫无疑问的。我们相信,在化学家和计算机科学家的努力下,这种全新的合成模式将使我们“手指点击合成”的梦想成为现实。
书本下载方式
关注公众号“新时代有机化学”,发送“分子科学中的机器学习”自动获取下载链接