放在最前面的参考链接:给学徒的ATAC-seq数据实战(附上收费视频)
数据来源的文章:
-
The landscape of accessible chromatin in mammalian preimplantation embryos. Nature 2016 Jun 30;534(7609):652-7. PMID: 27309802
 image.png
image.png
数据的GEO号:GSE66581
- 链接:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66581
- 健明说: 在SRA数据库可以下载原始测序数据 , 从文章找到数据的ID: https://www.ncbi.nlm.nih.gov/sra?term=SRP055881 把下面的内容保存到文件,命名为 srr.list 就可以使用prefetch这个函数来下载。
配置环境之软件的安装,这里首推通过conda来创建一个project专属的环境
- 可以无脑复制下面这段代码
# https://mirrors.tuna.tsinghua.edu.cn/help/anaconda/
# https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/
wget https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh
source ~/.bashrc
## 安装好conda后需要设置镜像。
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda
conda config --set show_channel_urls yes
conda create -n atac -y python=2 bwa
conda info --envs
source activate atac
# 可以用search先进行检索
conda search trim_galore
## 保证所有的软件都是安装在 wes 这个环境下面
conda install -y sra-tools
conda install -y trim-galore samtools bedtools
conda install -y deeptools homer meme
conda install -y macs2 bowtie bowtie2
conda install -y multiqc
conda install -y sambamba
数据的下载,由于原文数据太多,这里选取了四组数据来进行练习
- 创建文件config.sra内容如下(fastq-dump时候使用,以便将sra文件转换成fastq文件时候加上我们所需要的样品名称):
2-cell-1 SRR2927015
2-cell-2 SRR2927016
2-cell-5 SRR3545580
2-cell-4 SRR2927018
- 创建srr.list文件(里面为我们所需要下载的数据的SRR号)
SRR2927015
SRR2927016
SRR3545580
SRR2927018
数据下载
source activate atac
mkdir -p ~/project/atac/
cd ~/project/atac/
mkdir {sra,raw,clean,align,peaks,motif,qc}
cd sra
cat srr.list |while read id;do ( nohup prefetch $id & );done
## 默认下载目录:~/ncbi/public/sra/
- 下载完成后数据大小如下:
-rw-r--r-- 1 4.2G Nov 20 2015 SRR2927015.sra
-rw-r--r-- 1 5.5G Nov 20 2015 SRR2927016.sra
-rw-r--r-- 1 2.0G Nov 20 2015 SRR2927018.sra
-rw-r--r-- 1 7.0G May 20 2016 SRR3545580.sra
第一步:将sra文件转换成fastq文件
mv ~/ncbi/public/sra/SRR* sra/
- 创建sh文件01_fastq-dump.sh,内容如下:
## 下面需要用循环
## cd ~/project/atac/
source activate atac
dump=fastq-dump
analysis_dir=raw
# mkdir -p $analysis_dir # 由于之前已经创建过了,所以这里就无需创建了
## 下面用到的 config.sra 文件,就是上面自行制作的。
# $fastq-dump sra/SRR2927015.sra --gzip --split-3 -A 2-cell-1 -O clean/
cat config.sra |while read id;
do echo $id
arr=($id) # 这里可以类似看成获得矩阵
srr=${arr[1]} # 这里表示提取矩阵的第二列,即SRR号
sample=${arr[0]} # 这里表示提取矩阵的第一列,即样本名称
# 测序数据的sra转fasq
nohup $dump -A $sample -O $analysis_dir --gzip --split-3 sra/$srr.sra &
done
- 然后运行, 有集群的切勿在登陆节点运行,要切换到计算节点(不知道为什么我自己的不能通过提交任务qsub来运行)
sh 01_fastq-dump.sh
第二步,测序数据的过滤
- 手动创建一个包含fastq文件路径的config.raw文本文件,第一列随意填,充数,第二列为fastq1的路径,第二列为fastq2的路径。
1 ~/project/atac/raw/2-cell-1_1.fastq.gz ~/project/atac/raw/2-cell-1_2.fastq.gz
2 ~/project/atac/raw/2-cell-2_1.fastq.gz ~/project/atac/raw/2-cell-2_2.fastq.gz
3 ~/project/atac/raw/2-cell-4_1.fastq.gz ~/project/atac/raw/2-cell-4_2.fastq.gz
4 ~/project/atac/raw/2-cell-5_1.fastq.gz ~/project/atac/raw/2-cell-5_2.fastq.gz
- 创建02_trim_galore.sh文件,内容如下
cd ~/project/atac/
# mkdir -p clean
source activate atac
# trim_galore -q 25 --phred33 --length 35 -e 0.1 --stringency 4 --paired -o clean/ raw/2-cell-1_1.fastq.gz raw/2-cell-1_2.fastq.gz
cat config.raw |while read id;
do echo $id
arr=($id)
fq2=${arr[2]}
fq1=${arr[1]}
sample=${arr[0]}
nohup trim_galore -q 25 --phred33 --length 35 -e 0.1 --stringency 4 --paired -o clean $fq1 $fq2 &
done
ps -ef |grep trim
第三步,数据质量的检测
- 创建03_fastqc_multiqc.sh文本文件,内容如下
mkdir -p qc
cd ~/JMJ705_ChIP-seq/qc
mkdir -p clean
fastqc -t 5 ../clean/*gz -o clean/
mkdir -p raw
fastqc -t 5 ../raw/*gz -o raw/
- 使用multiqc进行合并质检结果
cd raw/
multiqc ./*zip
cd clean/
multiqc ./*zip
第四步,比对
- 健明说:比对需要的index,看清楚物种,根据对应的软件来构建,这里直接用bowtie2进行比对和统计比对率, 需要提前下载参考基因组然后使用命令构建索引,或者直接就下载索引文件:下载小鼠参考基因组的索引和注释文件, 这里用常用的mm10
- 下载索引文件
# 索引大小为3.2GB, 不建议自己下载基因组构建,可以直接下载索引文件,代码如下:
mkdir referece && cd reference
wget -4 -q ftp://ftp.ccb.jhu.edu/pub/data/bowtie2_indexes/mm10.zip
unzip mm10.zip
- 解压后文件大小
-rw-r--r-- 1 848M May 3 2012 mm10.rev.1.bt2
-rw-r--r-- 1 633M May 3 2012 mm10.rev.2.bt2
-rw-r--r-- 1 848M May 2 2012 mm10.1.bt2
-rw-r--r-- 1 633M May 2 2012 mm10.2.bt2
-rw-r--r-- 1 6.0K May 2 2012 mm10.3.bt2
-rw-r--r-- 1 633M May 2 2012 mm10.4.bt2
- 取少量数据进行比对,测试流程
zcat clean/2-cell-1_1_val_1.fq.gz |head -10000 > test_file/test1.fq
zcat clean/2-cell-1_2_val_2.fq.gz |head -10000 > test_file/test2.fq
bowtie2 -x ~/project/atac/referece/mm10 -1 test1.fq -2 test2.fq | samtools sort -@ 5 -O bam -o test.bam
# 三种不同的去重复软件
# 这里选用sambamba来去重复
sambamba markdup -r test.bam test.sambamba.rmdup.bam
samtools flagstat test.sambamba.rmdup.bam
samtools flagstat test.sambamba.rmdup.bam
samtools flagstat test.bam
## 接下来只保留两条reads要比对到同一条染色体(Proper paired) ,还有高质量的比对结果(Mapping quality>=30)
## 顺便过滤 线粒体reads
samtools view -h -f 2 -q 30 test.sambamba.rmdup.bam |grep -v chrM| samtools sort -O bam -@ 5 -o - > test.last.bam
bedtools bamtobed -i test.last.bam > test.bed
- 测试无错误,可进行下一步分析, 创建04_bowtie2_align_sambamba_markdup.sh文件,基本copy健明的就行,改改文件路径,内容如下:
cd ~/project/atac/align
ls ~/project/atac/clean/*_1.fq.gz > 1
ls ~/project/atac/clean/*_2.fq.gz > 2
ls ~/project/atac/clean/*_2.fq.gz |cut -d"/" -f 8|cut -d"_" -f 1 > 0 ## 这里最好自己逐步分开运行一下检测0里面的结果是否是你的sample名称
paste 0 1 2 > config.clean ## 供mapping使用的配置文件
## 相对目录需要理解
bowtie2_index=~/project/atac/referece/mm10
## 一定要搞清楚自己的bowtie2软件安装在哪里,以及自己的索引文件在什么地方!!!
#source activate atac
cat config.clean |while read id;
do echo $id
arr=($id)
fq2=${arr[2]}
fq1=${arr[1]}
sample=${arr[0]}
## 比对过程15分钟一个样本
bowtie2 -p 5 --very-sensitive -X 2000 -x $bowtie2_index -1 $fq1 -2 $fq2 |samtools sort -O bam -@ 5 -o - > ${sample}.raw.bam
samtools index ${sample}.raw.bam
bedtools bamtobed -i ${sample}.raw.bam > ${sample}.raw.bed
samtools flagstat ${sample}.raw.bam > ${sample}.raw.stat
# https://github.com/biod/sambamba/issues/177
sambamba markdup --overflow-list-size 600000 --tmpdir='./' -r ${sample}.raw.bam ${sample}.rmdup.bam
samtools index ${sample}.rmdup.bam
## ref:https://www.biostars.org/p/170294/
## Calculate %mtDNA:
mtReads=$(samtools idxstats ${sample}.rmdup.bam | grep 'chrM' | cut -f 3)
totalReads=$(samtools idxstats ${sample}.rmdup.bam | awk '{SUM += $3} END {print SUM}')
echo '==> mtDNA Content:' $(bc <<< "scale=2;100*$mtReads/$totalReads")'%'
samtools flagstat ${sample}.rmdup.bam > ${sample}.rmdup.stat
samtools view -h -f 2 -q 30 ${sample}.rmdup.bam |grep -v chrM |samtools sort -O bam -@ 5 -o - > ${sample}.last.bam
samtools index ${sample}.last.bam
samtools flagstat ${sample}.last.bam > ${sample}.last.stat
bedtools bamtobed -i ${sample}.last.bam > ${sample}.bed
done
其中bowtie2比对加入了-X 2000 参数,是最大插入片段,宽泛的插入片段范围(10-1000bp)
得到的bam文件如下:
-rw-r--r-- 1 523M Oct 12 07:15 ./2-cell-5.last.bam
-rw-r--r-- 1 899M Oct 12 07:14 ./2-cell-5.rmdup.bam
-rw-r--r-- 1 5.5G Oct 12 06:51 ./2-cell-5.raw.bam
-rw-r--r-- 1 427M Oct 12 03:23 ./2-cell-4.last.bam
-rw-r--r-- 1 586M Oct 12 03:23 ./2-cell-4.rmdup.bam
-rw-r--r-- 1 1.8G Oct 12 03:17 ./2-cell-4.raw.bam
-rw-r--r-- 1 678M Oct 12 02:22 ./2-cell-2.last.bam
-rw-r--r-- 1 1.1G Oct 12 02:20 ./2-cell-2.rmdup.bam
-rw-r--r-- 1 4.6G Oct 12 02:00 ./2-cell-2.raw.bam
-rw-r--r-- 1 490M Oct 11 23:02 ./2-cell-1.last.bam
-rw-r--r-- 1 776M Oct 11 23:01 ./2-cell-1.rmdup.bam
-rw-r--r-- 1 3.7G Oct 11 22:48 ./2-cell-1.raw.bam
- 上述脚本的步骤都可以拆分运行,比如bam文件构建index或者转为bed的:
ls *.last.bam|xargs -i samtools index {}
ls *.last.bam|while read id;do (bedtools bamtobed -i $id >${id%%.*}.bed) ;done
ls *.raw.bam|while read id;do (nohup bedtools bamtobed -i $id >${id%%.*}.raw.bed & ) ;done
- 最后得到的bed文件是
-rw-r--r-- 1 254M Oct 12 07:16 ./2-cell-5.bed
-rw-r--r-- 1 203M Oct 12 03:24 ./2-cell-4.bed
-rw-r--r-- 1 338M Oct 12 02:23 ./2-cell-2.bed
-rw-r--r-- 1 237M Oct 11 23:03 ./2-cell-1.bed
第五步,使用macs2找peaks
# macs2 callpeak -t 2-cell-1.bed -g mm --nomodel --shift -100 --extsize 200 -n 2-cell-1 --outdir ../peaks/
cd ~/project/atac/peaks/
ls *.bed | while read id ;do (macs2 callpeak -t $id -g mm --nomodel --shift -100 --extsize 200 -n ${id%%.*} --outdir ./) ;done
- macs2软件说明书详见:https://www.jianshu.com/p/21e8c51fca23
- 得到如下结果
-rw-r--r-- 1 1.1M Oct 12 14:35 2-cell-5_peaks.narrowPeak
-rw-r--r-- 1 690K Oct 12 14:35 2-cell-5_summits.bed
-rw-r--r-- 1 1.2M Oct 12 14:35 2-cell-5_peaks.xls
-rw-r--r-- 1 368K Oct 12 14:35 2-cell-4_peaks.narrowPeak
-rw-r--r-- 1 418K Oct 12 14:35 2-cell-4_peaks.xls
-rw-r--r-- 1 247K Oct 12 14:35 2-cell-4_summits.bed
-rw-r--r-- 1 1.2M Oct 12 14:35 2-cell-2_peaks.narrowPeak
-rw-r--r-- 1 1.4M Oct 12 14:35 2-cell-2_peaks.xls
-rw-r--r-- 1 805K Oct 12 14:35 2-cell-2_summits.bed
-rw-r--r-- 1 634K Oct 12 14:34 2-cell-1_peaks.narrowPeak
-rw-r--r-- 1 720K Oct 12 14:34 2-cell-1_peaks.xls
-rw-r--r-- 1 425K Oct 12 14:34 2-cell-1_summits.bed
第六步,计算插入片段长度,FRiP值,IDR计算重复情况
- 非冗余非线粒体能够比对的fragment、比对率、NRF、PBC1、PBC2、peak数、无核小体区NFR、TSS富集、FRiP 、IDR重复的一致性!
- 名词解释:https://www.encodeproject.org/data-standards/terms/
- 参考:https://www.encodeproject.org/atac-seq/
统计indel插入长度的分布
- 看 bam文件第9列,在R里面统计绘图 bam文件详解
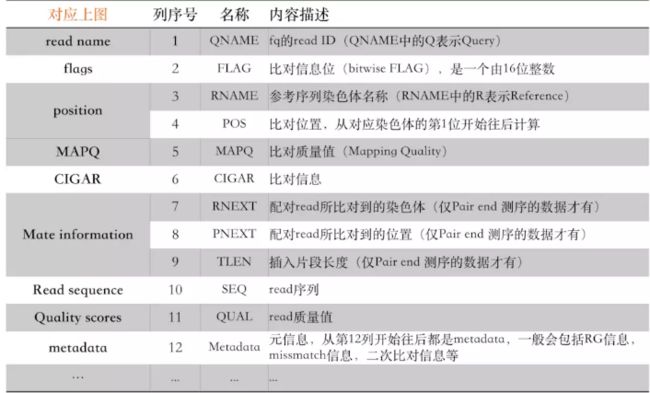 image.png
image.png - 提取bam文件的第九列, 创建一个config.last_bam文件,里面内容包含bam文件的名称
2-cell-1.last.bam 2-cell-1.last
2-cell-2.last.bam 2-cell-2.last
2-cell-4.last.bam 2-cell-4.last
2-cell-5.last.bam 2-cell-5.last
- 然后创建了提取bam文件的第九列indel插入长度信息的sh文件 indel_length.sh,内容如下:
cat config.last_bam |while read id;
do
arr=($id)
sample=${arr[0]}
sample_name=${arr[1]}
samtools view $sample |awk '{print $9}' > ${sample_name}_length.txt
done
- 然后我们得到四个bam文件的indel插入长度信息
-rw-r--r-- 1 24M Oct 12 15:27 2-cell-5.last_length.txt
-rw-r--r-- 1 19M Oct 12 15:27 2-cell-4.last_length.txt
-rw-r--r-- 1 32M Oct 12 15:26 2-cell-2.last_length.txt
-rw-r--r-- 1 22M Oct 12 15:26 2-cell-1.last_length.txt
cmd=commandArgs(trailingOnly=TRUE);
input=cmd[1]; output=cmd[2];
a=abs(as.numeric(read.table(input)[,1]));
png(file=output);
hist(a,
main="Insertion Size distribution",
ylab="Read Count",xlab="Insert Size",
xaxt="n",
breaks=seq(0,max(a),by=10)
);
axis(side=1,
at=seq(0,max(a),by=100),
labels=seq(0,max(a),by=100)
);
dev.off()
- 准备一个用于R语言批量绘制indel分布的文本输入文件config.indel_length_distribution
2-cell-1.last_length.txt 2-cell-1.last_length
2-cell-2.last_length.txt 2-cell-2.last_length
2-cell-4.last_length.txt 2-cell-4.last_length
2-cell-5.last_length.txt 2-cell-5.last_length
- 有了上面的文件就可以批量检验bam文件进行出图。创建批量运行的shell脚本indel_length_distribution.sh
cat config.indel_length_distribution |while read id;
do
arr=($id)
input=${arr[0]}
output=${arr[1]}
Rscript indel_length_distribution.R $input $output
done

image.png
FRiP值的计算:fraction of reads in called peak regions
- 单个举例
bedtools intersect -a 2-cell-1.bed -b 2-cell-1_peaks.narrowPeak |wc -l
148210
wc -l 2-cell-1.bed
5105850
# 故2-cell-1的FRiP为
148210/5105850 = 0.0292
- 批量计算 FRiP, 创建sh文件01_FRiP.sh
cd ~/project/atac/peaks
ls *narrowPeak|while read id;
do
echo $id
bed=$(basename $id "_peaks.narrowPeak").bed
#ls -lh $bed
Reads=$(bedtools intersect -a $bed -b $id |wc -l|awk '{print $1}')
totalReads=$(wc -l $bed|awk '{print $1}')
echo $Reads $totalReads
echo '==> FRiP value:' $(bc <<< "scale=2;100*$Reads/$totalReads")'%'
done
- 运行结果为
$ sh 01_FRiP.sh
2-cell-1_peaks.narrowPeak
148210 5105850
==> FRiP value: 2.90%
2-cell-2_peaks.narrowPeak
320407 7292154
==> FRiP value: 4.39%
2-cell-4_peaks.narrowPeak
90850 4399720
==> FRiP value: 2.06%
2-cell-5_peaks.narrowPeak
258988 5466482
==> FRiP value: 4.73%
- 健明记录:
Fraction of reads in peaks (FRiP) - Fraction of all mapped reads that fall into the called peak regions, i.e. usable reads in significantly enriched peaks divided by all usable reads. In general, FRiP scores correlate positively with the number of regions. (Landt et al, Genome Research Sept. 2012, 22(9): 1813–1831)
文章其它指标:https://www.nature.com/articles/sdata2016109/tables/4
可以使用R包看不同peaks文件的overlap情况
- 注意这里由于R版本是3.5.1,所以需要GCC版本要大于等于4.8,由于本服务器系统版本为4.7,所以需更改GCC版本,通过module命令调用更高的GCC版本
- 装包不成功,暂时不管
source /public/home/software/.bashrc
module load GCC/5.4.0-2.26
source activate atac
- 将narrowPeak文件传入到本地,使用本地R进行可视化
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
source("http://bioconductor.org/biocLite.R")
library('BiocInstaller')
# biocLite("ChIPpeakAnno")
# biocLite("ChIPseeker")
library(ChIPseeker)
library(ChIPpeakAnno)
setwd("E://desktop/sept/ATAC-seq_practice/find_peaks_overlaping/")
list.files('./',"*.narrowPeak")
tmp = lapply(list.files('./',"*.narrowPeak"),function(x){
return(readPeakFile(file.path('./', x)))
})
tmp
ol <- findOverlapsOfPeaks(tmp[[1]],tmp[[2]]) # 这里选取的是第一个文件和第二个文件,即cell.1_peak_1和cell.2_peak
png('overlapVenn.png')
makeVennDiagram(ol)
dev.off()
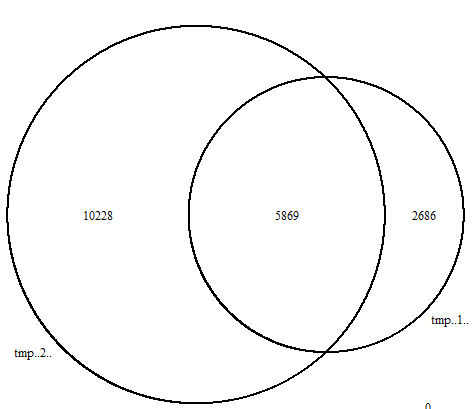
image.png
- 也可以使用专业软件,IDR 来进行计算出来,同时考虑peaks间的overlap,和富集倍数的一致性
conda create -n py3 -y python=3 idr
conda activate py3
idr -h
idr --samples 2-cell-1_peaks.narrowPeak 2-cell-2_peaks.narrowPeak --plot
第七步,deeptools的可视化
- 具体见:https://mp.weixin.qq.com/s/a4qAcKE1DoukpLVV_ybobA 在ChiP-seq 讲解。
- 首先把bam文件转为bw文件,详情:http://www.bio-info-trainee.com/1815.html
cd ~/project/atac/deeptools_result
#source activate atac # 由于原本电脑存在deeptools所以就没必要激活了
#ls *.bam |xargs -i samtools index {}
ls *last.bam |while read id;do
nohup bamCoverage -p 5 --normalizeUsingRPKM -b $id -o ${id%%.*}.last.bw &
done
# cd dup
# ls *.bam |xargs -i samtools index {}
# ls *.bam |while read id;do
# nohup bamCoverage --normalizeUsing CPM -b $id -o ${id%%.*}.rm.bw &
# done
- mm10的Refgene文件的下载,
- 第一种参考链接:ChIP-seq基础入门学习
curl 'http://genome.ucsc.edu/cgi-bin/hgTables?hgsid=646311755_P0RcOBvAQnWZSzQz2fQfBiPPSBen&boolshad.hgta_printCustomTrackHeaders=0&hgta_ctName=tb_ncbiRefSeq&hgta_ctDesc=table+browser+query+on+ncbiRefSeq&hgta_ctVis=pack&hgta_ctUrl=&fbQual=whole&fbUpBases=200&fbExonBases=0&fbIntronBases=0&fbDownBases=200&hgta_doGetBed=get+BED' >mm10.refseq.bed
- 第二种:从http://genome.ucsc.edu/cgi-bin/hgTables下载
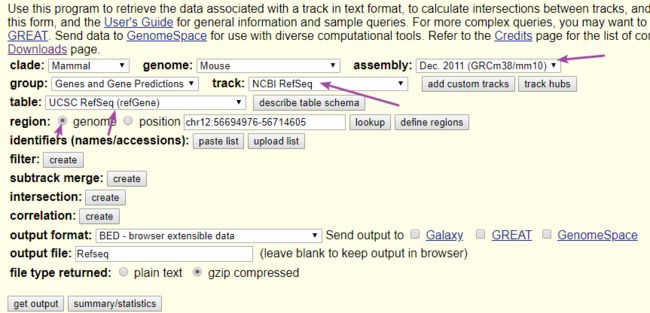 image.png
image.png - 这里选取了第二种方法得到的bed文件进行后续可视化
查看TSS附件信号强度:创建07_deeptools_TSS.sh
- depptools 使用说明
## both -R and -S can accept multiple files
mkdir -p ~/project/atac/tss
cd ~/project/atac/tss
# source activate atac # 由于我这里自己系统有就没调用了
computeMatrix reference-point --referencePoint TSS -p 15 \
-b 10000 -a 10000 \
-R ~/project/atac/mm10_Refgene/Refseq.bed \
-S ~/project/atac/deeptools_result/*.bw \
--skipZeros -o matrix1_test_TSS.gz \
--outFileSortedRegions regions1_test_genes.bed
## both plotHeatmap and plotProfile will use the output from computeMatrix
plotHeatmap -m matrix1_test_TSS.gz -out test_Heatmap.png
plotHeatmap -m matrix1_test_TSS.gz -out test_Heatmap.pdf --plotFileFormat pdf --dpi 720
plotProfile -m matrix1_test_TSS.gz -out test_Profile.png
plotProfile -m matrix1_test_TSS.gz -out test_Profile.pdf --plotFileFormat pdf --perGroup --dpi 720
-
出图展示
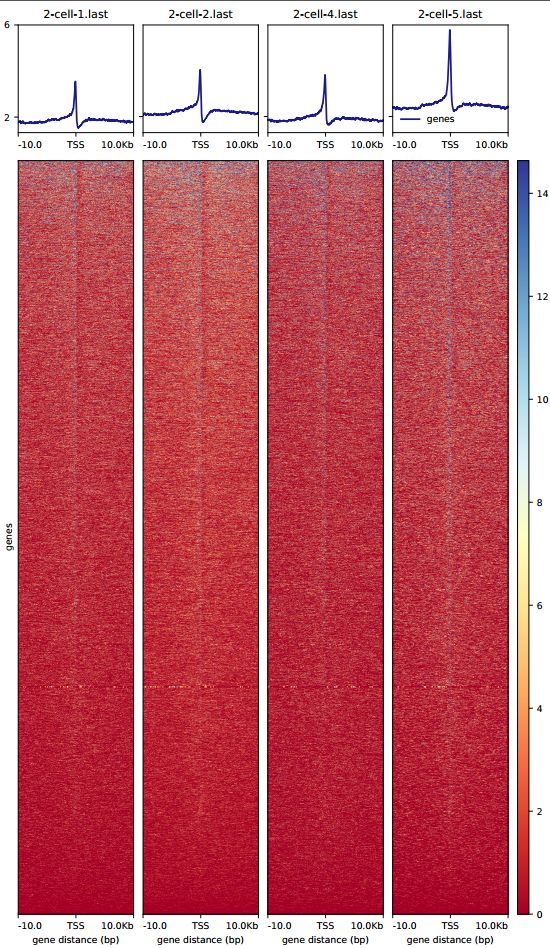 image.png
image.png
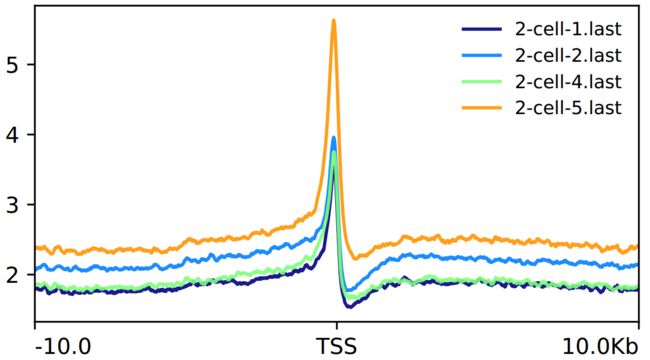 image.png
image.png
 image.png
image.png - 查看基因body的信号强度,创建07_deeptools_Body.sh
#source activate atac
mkdir Body
cd ~/project/atac/Body
computeMatrix scale-regions -p 15 \
-R ~/project/atac/mm10_Refgene/Refseq.bed \
-S ~/project/atac/deeptools_result/*.bw \
-b 10000 -a 10000 \
--skipZeros -o matrix1_test_body.gz
# plotHeatmap -m matrix1_test_body.gz -out ExampleHeatmap1.png
plotHeatmap -m matrix1_test_body.gz -out test_body_Heatmap.png
plotProfile -m matrix1_test_body.gz -out test_body_Profile.png
plotProfile -m matrix1_test_body.gz -out test_Body_Profile.pdf --plotFileFormat pdf --perGroup --dpi 720
- 出图展示,注意下图可以看出body区明显的要短于两侧,如果要调整宽度,可自行调整以下参数
- --regionBodyLength
- --binSize
- 参考官网参数含义:https://deeptools.readthedocs.io/en/develop/content/tools/computeMatrix.html
image.png
 image.png
image.png
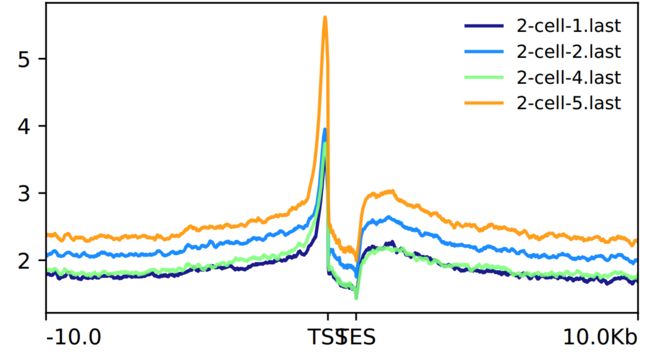 image.png
image.png
- ngsplot也是一个画profiler图的利器。
第八步:peaks注释
- 参考老版本教程链接:CS3: peak注释
- peaks区间注释分布
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
source("http://bioconductor.org/biocLite.R")
library('BiocInstaller')
biocLite("ChIPpeakAnno")
library(ChIPpeakAnno)
setwd("E://desktop/sept/ATAC-seq_practice/peaks_annotaion/")
biocLite("TxDb.Mmusculus.UCSC.mm10.knownGene")
biocLite("org.Mm.eg.db")
txdb <- TxDb.Mmusculus.UCSC.mm10.knownGene
promoter <- getPromoters(TxDb=txdb,
upstream=3000, downstream=3000)
files = list(cell_1_summits = "2-cell-1_summits.bed", cell_2_summits = "2-cell-2_summits.bed",
cell_4_summits = "2-cell-4_summits.bed", cell_5_summits = "2-cell-5_summits.bed")
peakAnno <- annotatePeak(files[[1]], # 分别改成2或者3或者4即可,分别对应四个文件
tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Hs.eg.db")
plotAnnoPie(peakAnno)
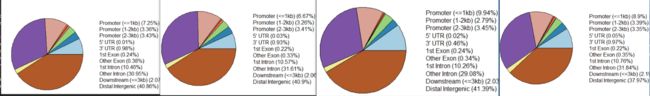
image.png
- plotAnnoBar来展示
plotAnnoBar(peakAnno)

image.png
- vennpie来展示
vennpie(peakAnno)

image.png
- upsetplot来展示
upsetplot(peakAnno)
upsetplot(peakAnno, vennpie=TRUE)
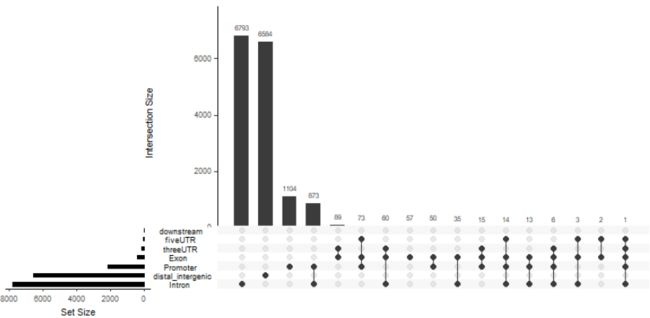
image.png
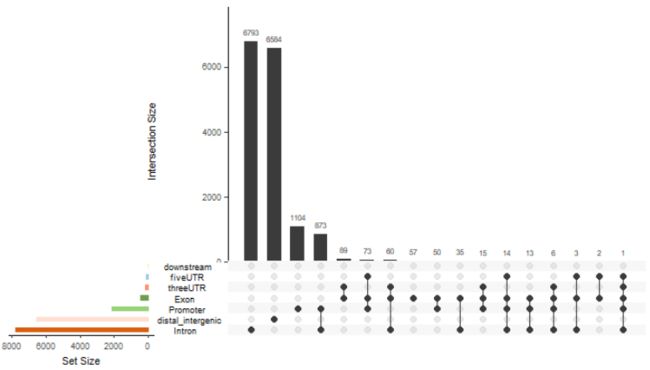
vennpie=TRUE
- ChIPseeker还注释了最近的基因,peak离最近基因的距离分布是什么样子的?ChIPseeker提供了plotDistToTSS函数来画这个分布:
plotDistToTSS(peakAnno,
title="Distribution of transcription factor-binding loci\nrelative to TSS")
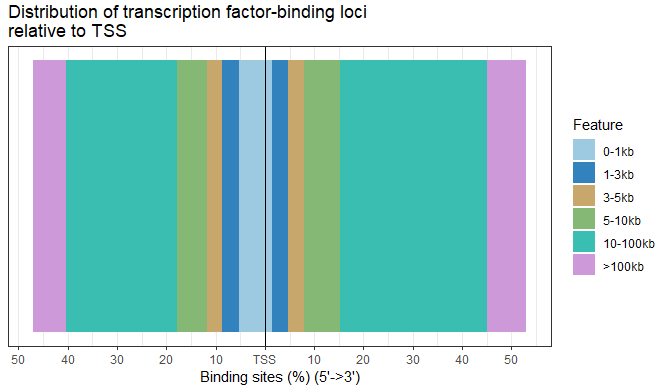
image.png
- plotAnnoBar和plotDistToTSS这两个柱状图都支持多个数据同时展示,方便比较,比如:
peakAnnoList <- lapply(files, annotatePeak,
TxDb=txdb,tssRegion=c(-3000, 3000))
plotAnnoBar(peakAnnoList)
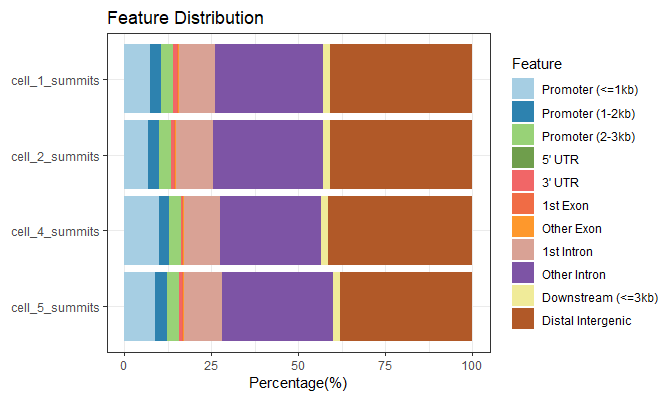
image.png
- 批量绘制距离TSS的百分比图
plotDistToTSS(peakAnnoList)
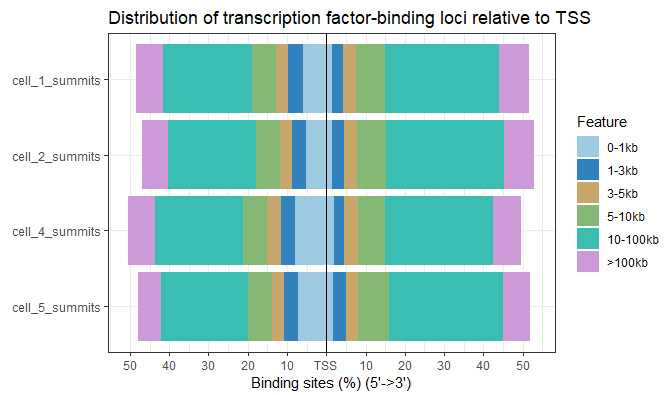
image.png
- ChIPseeker还提供了一个vennplot函数,比如我想看注释的最近基因在不同样本中的overlap:
genes <- lapply(peakAnnoList, function(i)
as.data.frame(i)$geneId)
vennplot(genes[2:4], by='Vennerable')
- ChIPseeker还提供了一个vennplot函数,比如我想看注释的最近基因在不同样本中的overlap:
genes <- lapply(peakAnnoList, function(i)
as.data.frame(i)$geneId)
vennplot(genes[2:4], by='Vennerable')
- 新版本ChIPpeakAnno()可视化后续再补上
- 参考链接:The ChIPpeakAnno user’s guide
 image.png
image.png
- 参考链接:The ChIPpeakAnno user’s guide
使用hommer进行注释
perl ~/miniconda3/envs/atac/share/homer-4.9.1-6/configureHomer.pl -install mm10 # 此不能再计算节点运行,需先在登陆节点运行下载
## 保证数据库下载是OK
# ls -lh ~/miniconda3/envs/atac/share/homer-4.9.1-5/data/genomes
# source activate atac
mkdir hommer_anno
cp peaks/*narrowPeak hommer_anno/
cd ~/project/atac/hommer_anno
ls *.narrowPeak |while read id;
do
echo $id
awk '{print $4"\t"$1"\t"$2"\t"$3"\t+"}' $id >${id%%.*}.homer_peaks.tmp
annotatePeaks.pl {id%%.*}.homer_peaks.tmp mm10 1>${id%%.*}.peakAnn.xls
2>${id%%.*}.annLog.txt
done
- 然后将peakAnn.xls文件导入到本地,通过excel透视功能进行可视化
- 用到的函数COUNTIF(), SUM()
- 这里还用到一个用来上传gif文件,生成链接的在线网页 http://thyrsi.com/
- 操作gif动图
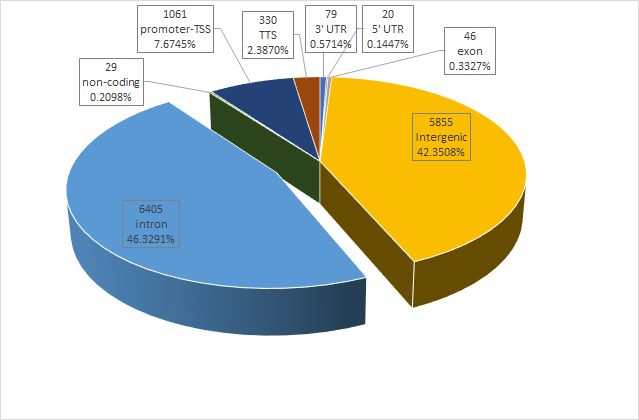
image.png
第九步,motif寻找及注释
- 创建08_hommer_motif.sh 文件
# mkdir -p ~/project/atac/motif
cd ~/project/atac/motif
# source activate atac
ls ../peaks/*.narrowPeak |while read id;
do
file=$(basename $id )
sample=${file%%.*}
echo $sample
awk '{print $4"\t"$1"\t"$2"\t"$3"\t+"}' $id > ${sample}.homer_peaks.tmp
nohup findMotifsGenome.pl ${sample}.homer_peaks.tmp mm10 ${sample}_motifDir -len 8,10,12 &
done
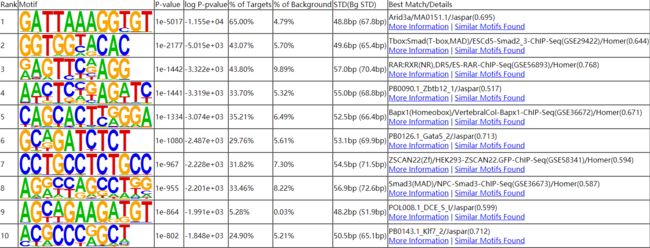
homerResults

knownResults
第十步,差异peaks分析
- diffbind
- DESeq2等后续进行分析
写在最后的话,通过此流程的实践,再次学到很多新的知识点以及脚本操作。
- 批量运行命令以及可视化
- 简单R脚本程序的编写