QIIME2进阶六_QIIME2训练分类器及物种注释
本文我们主要介绍了如何训练Naive Bayes分类器并把这个分类器应用于扩增子基因序列的物种注释与可视化。
本教程将使用来自人源化(humanized)小鼠的一组粪便样品,展示16S rRNA基因扩增子数据的“典型”QIIME 2分析。本教程旨在探讨人源化小鼠的遗传背景影响微生物群落的假设。然而,我们还需要考虑其他可能驱动微生物结构而不是小鼠基因型的混杂因素。
在本节中,我们将探索样本的物种组成情况。这个过程的第一步是为FeatureData[Sequence]的序列进行物种注释。我们将使用经过Naive Bayes分类器进行预训练,并通过q2-feature-classifier插件裁剪出符合样品制备和测序参数进行(即扩增的引物和测序序列的长度)训练分类器。我们将把这个分类器应用到序列中,并且可以生成从序列到物种注释结果关联的可视化。
01训练分类器 Training classifier
因为不同实验的扩增区域不同,鉴定物种分类的精度不同,提前的训练可以让分类结果更准确。在本教程中,使用到的是已经训练好细菌V4区的分类器,可在QIIME2官网下载。下面将以小鼠数据为例,详细解读分类器的训练方法。
| 方法注释在训练分类器之前,我们要弄清楚三点:1.选择合适的数据库;2.扩增引物;3测序序列长度分布(此步骤可选择使用)。 |
在本教程中,使用的16s rRNA 扩增子数据,引物为515f-806r(详细示例数据信息请看实战示例教程(1))。因此,选用greengenes数据库进行比对。
提取扩增区命令:
qiime feature-classifier extract-reads
--i-sequences 99_otus.qza
--p-f-primer ACTCCTACGGGAGGCAGCAG
--p-r-primer GGACTACHVGGGTWTCTAAT
--p-n-jobs 4
--o-reads ref-seqs.qza
仅保留细菌的代表序列命令:
qiime taxa filter-seqs
--i-sequences ref-seqs.qza
--i-taxonomy 99_otu_taxonomy.qza
--p-include Bacteria
--o-filtered-sequences ref-seqs-Bacteria.qza
仅保留细菌的注释信息命令:
qiime rescript filter-taxa
--i-taxonomy 99_otu_taxonomy.qza
--m-ids-to-keep-file ref-seqs-Bacteria.qza
--o-filtered-taxonomy ref-seqs-Bacteria-tax.qza
训练分类器命令:
qiime feature-classifier fit-classifier-naive-bayes
--i-reference-reads ref-seqs-Bacteria.qza
--i-reference-taxonomy ref-seqs-Bacteria-tax.qza
--o-classifier classifier-Bacteria.qza
| 方法注释:在完成提取扩增区后,可直接进行训练分类器。但由于部分数据库中同时含有细菌和古菌,有时还含有真核生物的特征序列和分类信息,在比对过程中可能会导致菌属比对错误等情况的出现,所以提取单一菌属序列及分类信息有助于提高比对的准确度。此步骤可以视测序数据实际情况而定,在处理真菌ITS数据时,不需要提取单一菌属。 |
02物种注释 Taxonomic classification
物种数据库
| 数据库名称 | 链接 | 说明 |
| RDP | http://rdp.cme.msu.edu/ | 一个辅助数据库,提供核糖体数据和有关的程序及服务。包括用于比对使用的rRNA序列。 |
| SILVA | http://www.arb-silva.de/ | 提供全面、质检和定期更新的数据用于比对细菌、古菌、Eukarya小(16S/18S,SSU)和大亚基(23S/28S,LSU)rRNA的所有序列。 |
| GreenGenes | htp://http://greengenes.secondgenome.com | 质控、全面的16S rRNA基因参考数据库。 |
| UNITE | https://unite.ut.ee | UNITE数据库是目前真菌ITS整理最全面的数据库,基于上百万的全长ITS高质量序列,包括45万多个假定物种。 |
本教程中物种注释使用已经训练好的GreenGene13.8的99%聚类序列的V4区分类器。该分类器通过识别对特定分类群体具有诊断性的k聚体,并使用该信息来预测每个特征的从属关系。
k聚体:即k-mer,将序列分成包含k个碱基的字符串。
物种注释命令:
qiime feature-classifier classify-sklearn
--i-reads dada2_rep_set.qza
--i-classifier gg-13-8-99-515-806-nb-classifier.qza
--o-classification taxonomy.qza
可视化物种注释命令:
qiime metadata tabulate
--m-input-file taxonomy.qza
--o-visualization taxonomy.qzv
输出结果文件:taxonomy.qza,即物种注释结果;taxonomy.qzv,物种注释表。
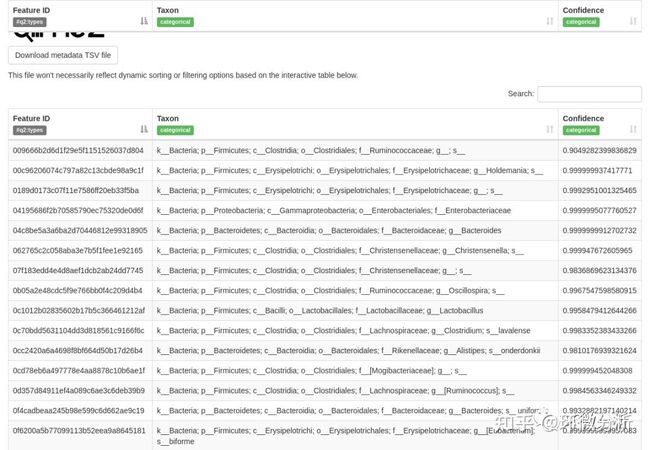
上表中包括界、门、纲、目、科、属和种的注释,可查看细菌物种信息和分类置信度。
生成物种丰度柱状图命令:
qiime taxa barplot
--i-table dada2-table.qza
--i-taxonomy taxonomy.qza
--m-metadata-file metadata.tsv
--o-visualization taxa-bar-plots.qzv
输出文件结果:taxa-bar-plots.qzv,即物种丰度柱状图。
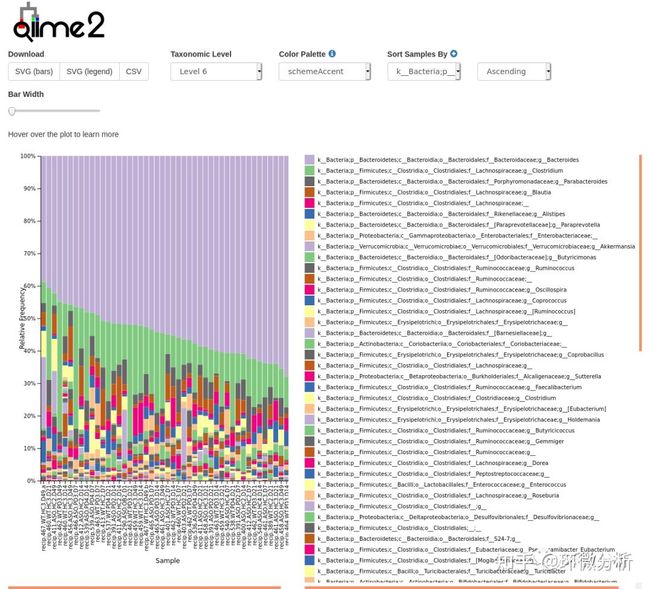
上图为细菌属水平样本堆叠柱状图。根据分类学分析结果,可以得知一个或多个样品在各分类学水平上的物种组成比例情况,反映不同样品在各分类学水平上的群落结构。taxa-bar-plots.qzv文件中可切换不同分类级别、选择10余种配色方案;切换排序类型和升降序方向。同时图中的柱可鼠标悬停查看数据。横坐标中每一个条形图代表一个样本,纵坐标代表该分类层级的序列数目或比例。同一种颜色代表相同的分类级别。图中的每根柱子中的颜色表示该样本在不同级别(门、纲、目等)的序列数目,序列数目只计算级别最低的分类,例如在属中计算过了,则在科中则不重复计算。
本文提供分析所需文件与所有输出结果文件,百度网盘下载链接:
https://pan.baidu.com/s/1sHhsOKeJuZrbgcfNGQ8X4Q
提取码:1234
这篇推文对你有帮助吗?喜欢这篇文章吗?喜欢就不要错过呀,关注本知乎号查看更多的环境微生物生信分析相关文章。亦可以用微信扫描下方二维码关注“环微分析”微信公众号,小编在里面载入了更加完善的学习资料供广大生信分析研究者爱好者参考学习,也希望读者们发现错误后予以指出,小编愿与诸君共同进步!!!

学习环境微生物分析,关注“环微分析”公众号,持续更新,开源免费,敬请关注!
转载自原创文章:
QIIME2进阶六_QIIME2训练分类器及物种注释
最后,再次感谢你阅读本篇文章,真心希望对你有所帮助。感谢!