本文转载自:https://www.jianshu.com/p/8b177773ae38
RNA-seq基本流程
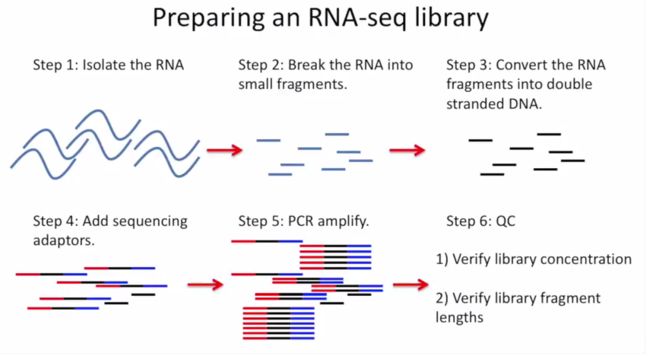
把RNA破碎成小片段,然后将RNA转变成一条cDNA,这一步需要用到反转录酶 reverse transcriptase (RT) 才能用RNA作为模板合成DNA。
不论是转录还是反转录都需要引物。通常如果我们要mRNA,那就可以用oligo-dT作为RT的引物,但是用它有两个问题,第一个是只能反转录那些有A尾巴的RNA,第二个问题是RT不是一个高度持续性的聚合酶,可能让转录提前发生终止,造成的结果就是3'端要比5'端reads富集,这样就会使得后续定量分析带来bias。
另一种常用的引物称为随机引物,随机引物的好处是没有A尾巴的诸如ncRNA也被留下了,而且不会存在明显的3'端偏差。但是很多研究也发现,所谓的随机引物根本就不随机,这也是测序结果中,通常前6个碱基的GC含量分布特别不均匀的原因。这几个碱基GC含量均匀很可能不是接头或者barcode那些东西,其实是Illumina 测序RT这一步的random hexamer priming 造成的bias,很多人在处理数据的时候会把这几个碱基去掉,其实很多时候真多RNA-seq数据去不去掉基本什么影响,不过开头如果有低质量的碱基倒是应该去掉。
随后是第二条链合成,这一步用是DNA聚合酶,以刚才和成的第一条链作为模板。
接下来就是在序列两端加上接头,加接头一方面是为了让机器可以识别这些序列,把这些序列固定;二是为了让多个样品可以同时上机,平摊每个样品的测序价格。双端测序为了让read从两边开始延伸,也需要在两端有所需的引物。
所谓双端测序,因为很多时候read的长度要短于insert,为了增加覆盖度于是就想出了从insert两端同时测序的办法。使得测序深度增加的同时也能够用来判断isoform方向。
对于illumina数据,有一条5-3的universal adaptor;还有一条是3-5的indexed adatpor,这条引物含有特意的barcode。需要说明的是,在双端测序中,如果insert 不是足够长,那么R1可能就会测到R2的引物,同时R2可能会测到R1引物的反向互补序列。
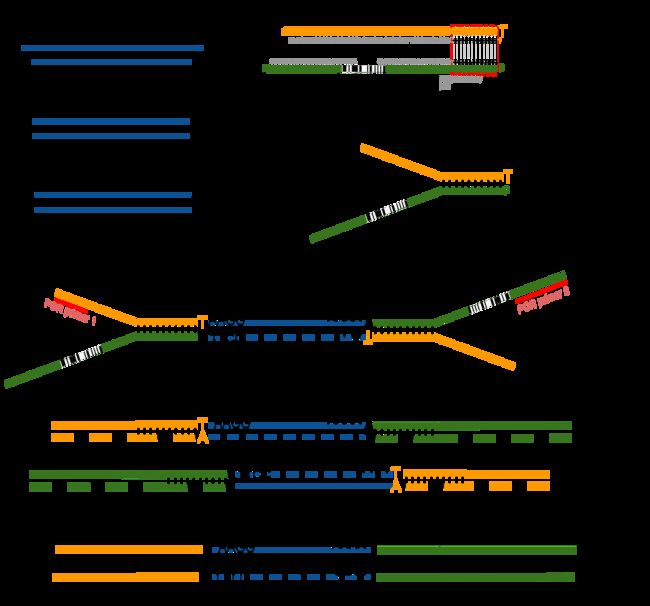
加了接头以后进行PCR的扩增。扩增后就开始测序,测序的过程如下图所示。
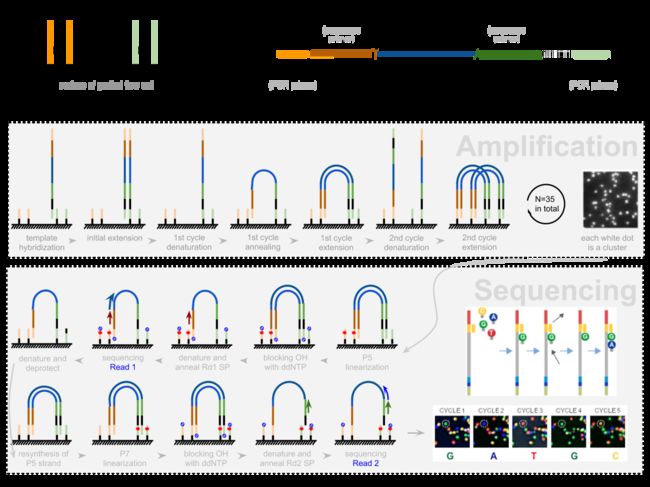

测序的基本思想是机器识别四种碱基发出的不同颜色的荧光,可以理解为一个flow cell 立着非常多序列,机器一层一层扫过去,通过识别荧光而判断这一层每个序列的碱基是什么。
链特异性测序
和普通的RNAseq不同,链特异性测序可以保留最初产生RNA的方向,普通建库方式为什么不行呢?因为传统建库方式通过两个接头的ligation把RNA已经变成了双链DNA,最后的文库中一部被测序的链对应正义链(sense strand),一部分被测序的链测是反义链。
链特异性建库方式有不止一种,对应到不同的软件又有不同的叫法,下面是几种称呼。要记住的是dUTP 测序方式的名字是fr-firstrand,也是RF。 至于具体的read方向接下来通过更详细的IGV截图说明问题。


链特异性建库方式(以目前最常用的dUTP为例,如下图所示)首先利用随机引物合成RNA的一条cDNA链,在合成第二条链的时候用dUTP代替dTTP,加adaptor后用UDGase处理,将有U的第二条cDNA降解掉。
https://upload-images.jianshu.io/upload_images/177622-d2cee6b7279efb66.png?imageMogr2/auto-orient/strip|imageView2/2/format/webp
这样最后的insert DNA fragment都是来自于第一条cDNA,也就是dUTP叫fr-firststrand的原因。对于dUTP数据,tophat的参数应该为–library-type fr-firststrand。这里的first-strand cDNA可不是RNA strand,在使用htseq-count 时,真正的正义链应该是使用参数-s reverse 得到的结果。
正正反反不清楚
说到链特异性测序,实在让人困惑的是各种链的概念,尤其是翻译成中文就更说不清了。
DNA 的正链和负链,就是那两条反向互补的链。参考基因组给出的那个链就是所谓的正链(forword),另一条链是反链(reverse)。但是这正反一定不能和正义链(sense strand)反义链(antisense strand)混淆,两条互补的DNA链其中一条携带编码蛋白质信息的链称为正义链,另一条与之互补的称为反义链。但是携带编码信息的正义链不是模板,只是因为它的序列和RNA相同,正义链也是编码链。而反义链虽然和RNA反向互补,但它可是真正给RNA当模板的链,因此反义链也是模板链。
总结两点
1、正义链(sense strand)= 编码链(coding strand)= 非模板链
2、forword strand 上可以同时有sense strand 和 antisense strand。因为这完全是两个不同的概念。
dUTP到底是怎么回事
从前文的一个图我们可以总结出dUTP方式测序R1文件中read1 的方向和基因的方向(正义链)是相反的,而R2文件中的read2 方向和基因的方向是相同的。
可以参考下面的两个igv文件bam截图。
下面这个图示按照igv 颜色选项中的read strand 方向进行区分,可以看到所有红色read都是在正链方向(注意正链不是正义链),而所有蓝色的read都是负链方向。下面基因的方向是正链方向,也就是和粉色的read同向的,如果你把鼠标放到随意一个粉色的read上,就能看到显示的信息是second of pair,也就是pair中的read2(R2);反之如果你在蓝色的read上面,就会显示信息是first of pair,也就是R1 。
总结,dUTP测序中pair read 中的read1(R1)和基因方向相反,read2(R2)和基因方向相同。
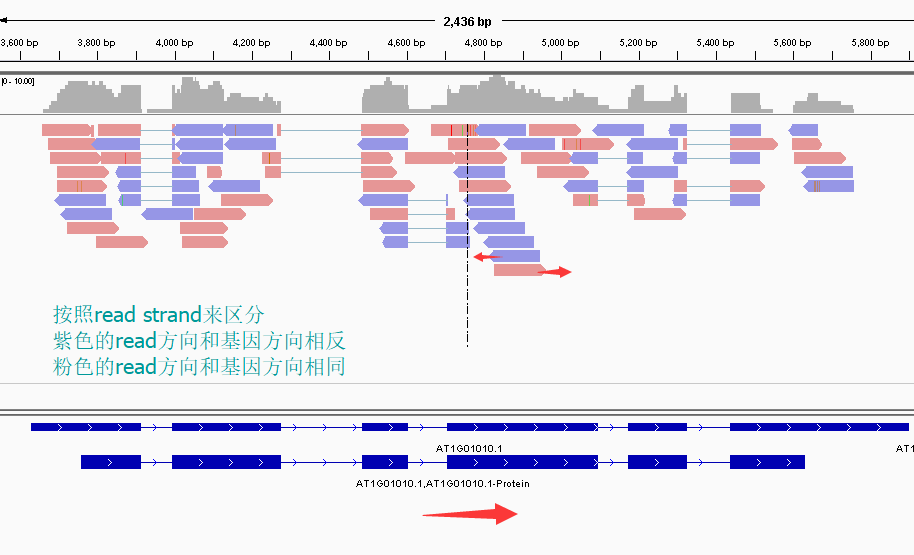
再看下面这张图
https://upload-images.jianshu.io/upload_images/177622-8d88e440355bf719.png?imageMogr2/auto-orient/strip|imageView2/2/format/webp
这张图展示了两个基因1和2,我们可以发现gene1的正义链就在正链上,而gene2的正义链其实是在反链上。看read情况,a,c两个read虽然针对正链负链而言方向一致,都是负链方向,但是如果把a是pair中的read1(first of pair ),而c是pair中的read2(second of pair)。也就是说,read方向一致,但一个是read1一个是read2,说明这两个read对应的基因一定是反向的。同样的道理,虽然b,d都是两个方向为负链的read,但是b其实是所在pair的read2(second of pair),而d是所在pair的read1(first of pair)。
再次强调,dUTP测序中pair read 中的read1(R1)和基因方向相反,read2和基因方向相同
当使用read strand来进行颜色区分的时候,每一个基因上两种颜色的分布应该相对均匀(也就是所谓的pair end)。
如果这个时候把颜色选项改为按照first of pair of strand来区分,会出现下图的变化。
https://upload-images.jianshu.io/upload_images/177622-37a185b87324569b.png?imageMogr2/auto-orient/strip|imageView2/2/format/webp
geng1的read全部变成了紫色,而gene2的read全部变成了粉色。
如果是非链特异性测序,在first of pair of strand模式下,同一个gene相关的read颜色也是明显混杂的。如下图:
https://upload-images.jianshu.io/upload_images/177622-fa296437d1301175.png?imageMogr2/auto-orient/strip|imageView2/2/format/webp
几个常用软件的设置
STAR mpping 时无需特别设置,但如果不是链特异性数据且下游分析要用到cufflinks 则需要增加参数 --outSAMstrandField intronMotif。为的是增加一个XS标签。
If you have stranded RNA-seq data, you do not need to use any specific STAR options. Instead, you need to run Cufflinks with the library option --library-type options. For example, cufflinks... --library-type fr-firststrand should be used for the standard dUTP protocol, including Illumina’s stranded Tru-Seq.
hisat2 --rna-strandness RF
目的也是给比对序列添加一个XS标签以区分方向,方面cufflinks使用。
For single-end reads, use F or R. 'F' means a read corresponds to a transcript. 'R' means a read corresponds to the reverse complemented counterpart of a transcript. For paired-end reads, use either FR or RF.
With this option being used, every read alignment will have an XS attribute tag: '+' means a read belongs to a transcript on '+' strand of genome. '-' means a read belongs to a transcript on '-' strand of genome.