Graph Isomorphism Network(GIN)
在上一节GraphSAGE中,我们牺牲了部分准确率来增强模型的可伸缩性。在本节中,我们将引入Weisfeiler-Leman(WL)检验的概念,该理念可以帮助我们更好的表达GNNs架构。
文章目录
- 前言
- 一、WL检验原理
- 二、GIN原理
- 三、最大池化层
- 四、GIN的应用
-
- 1、Proteins数据集
- 2、预处理数据集
- 3、类函数定义
- 4、模型训练
- 总结
前言
神经网络在过去被用于逼近函数,具有一层隐藏层的神经网络前馈神经网络可以逼近任何平滑函数。那么如何近似图架构下的通用函数呢?
在GNNs架构下,我们的目标是尽可能产生有区分度的嵌入向量。这既代表不同的节点有不同的嵌入向量,同时相似的节点其对应的嵌入向量也具有相似性。为了计算嵌入向量,我们将节点特征和其邻接节点相联系。
在图论中,这被称为图同构问题,如果两个图是同构的,那么它们唯一的区别就在于节点排列的不同。图1体现了两个同构图。

一、WL检验原理
WL检验的工作流程如下:
- 首先,给所有节点赋予相同的颜色
- 每一个节点聚合自己的颜色和邻接节点的颜色
- 合成的颜色输入到哈希函数中产生新的颜色
- 每一个节点聚合自己的新颜色和邻接节点的新颜色
- 合成的新颜色再次输入到哈希函数中产生新的颜色
- 不断重复上述步骤,直到所有节点都不在发生颜色变化
图2可以帮助我们理解WL检验的流程。
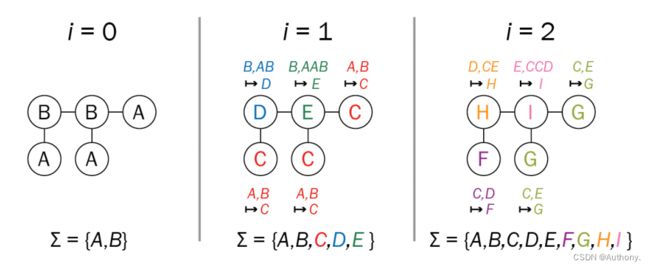
在检验之后,如果两图不同的颜色则两图非同构,否则则不能确定两图是否同构。
上述同构的步骤与GNN有异曲同工之妙,颜色作为嵌入向量的一种形式,哈希函数是一个聚合器,但是这个聚合器不同与我们在GNN中所采用的平均值聚合或者最大值聚合。哈希函数所用的是累加聚合。
- 在平均值聚合下,邻接节点有10个红色和10个蓝色同邻接节点只有1个红色和1个蓝色相同。
- 在最大值聚合下,聚合器只会考虑蓝色和红色中的一个颜色。
- 在累加聚合下,邻接节点中有10个红色和10个蓝色异于1个红色和1个蓝色。
因此,累加聚合器相比于另外两个聚合器可以区分更多的图架构。
二、GIN原理
GNN架构一般分为两个部分:
- 聚合(Aggregate):选择合适的邻接节点。
- 组合(Combine):将邻接节点的嵌入向量相组合形成目标节点的嵌入向量。
嵌入过程的表达式如下:
h i ′ = ϕ ( h i , f ( { h j : j ∈ N i } ) ) h_{i}^{\prime}=\phi\left(h_{i}, f\left(\left\{h_{j}: j \in \mathcal{N}_{i}\right\}\right)\right) hi′=ϕ(hi,f({hj:j∈Ni}))
在上式中, f f f函数用于聚合邻接节点, ϕ \phi ϕ用于应用选定的聚合器。在 G r a p h S A G E GraphSAGE GraphSAGE中,采样邻接节点为 f f f函数,而聚合器则为 m e a n mean mean、 L S T M LSTM LSTM和 m a x max max聚合。
在GIN中,为了将不同的输入内射到不同的输出中,引入了内射函数。
在GATs中,我们引入了自注意力权重。相类似的,我们首先利用多层感知机(MLP)嵌入向量,之后利用 R e L U ReLU ReLU对归一化后的嵌入向量进行处理。
h i ′ = R e L u ( N o r m ( M L P ( ( 1 + ε ) ⋅ h i + ∑ j ∈ N i h j ) ) ) h_{i}^{'}=\mathrm{Re}Lu\left( Norm\left( MLP\left( (1+\varepsilon )\cdot h_i+\sum_{j\in \mathcal{N} _i}{h_j} \right) \right) \right) hi′=ReLu Norm MLP (1+ε)⋅hi+j∈Ni∑hj
在上式中, ε \varepsilon ε是一个可学习的参量,代表了目标节点相较于邻接节点的重要性,同时多次重复上述步骤才能达到WL检验,即图2所示的效果。
三、最大池化层
图的分类依赖于嵌入向量的处理过程,这一步骤被称为全局池化(golbal pooling)或基于图水平特征提取(graph-level readout)。同邻接节点的处理相同,也有以下三种方法可以处理图的嵌入向量:
- 平均全局池化
- 最大全局池化
- 累加全局池化
在前文中,我们直到累加全局池化对于图的处理效果要优于其余两种。另外,为了考虑所有的结构信息,需要将WL检验过程中所产生的节点嵌入向量相连接。即:
h G = ∑ i = 0 N h i 0 ∥ ⋯ ∥ ∑ i = 0 N h i k h_G=\left. \sum_{i=0}^N{h_{i}^{0}} \right\| \cdots \left\| \sum_{i=0}^N{h_{i}^{k}} \right. hG=i=0∑Nhi0 ⋯ i=0∑Nhik
四、GIN的应用
1、Proteins数据集
该数据集包含1113个表示蛋白质的图,其中每个节点都是一个氨基酸。当两个节点的距离小于0.6纳米时,一条边连接两个节点。该数据集的目标是判断每种蛋白质是否为酶。酶是一种特殊类型的蛋白质,它起到催化剂的作用,加速细胞中的化学反应。例如,一种叫做脂肪酶的酶有助于食物的消化。图3显示了蛋白质的三维图。
首先,我们将数据集导入。
import torch
import torch.nn.functional as F
import numpy as np
import matplotlib.pyplot as plt
import networkx as nx
from collections import Counter
from torch_geometric.utils import to_networkx
from torch.nn import Linear, Sequential, BatchNorm1d, ReLU, Dropout
from torch_geometric.nn import GCNConv, GINConv
from torch_geometric.nn import global_mean_pool, global_add_pool
from torch_geometric.datasets import TUDataset
from torch_geometric.loader import DataLoader
torch.manual_seed(0)
torch.cuda.manual_seed(0)
torch.cuda.manual_seed_all(0)
torch.backends.cudnn.deterministic = True
torch.backends.cudnn.benchmark = False
dataset = TUDataset(root='.', name='PROTEINS').shuffle()
# Print information about the dataset
print(f'Dataset: {dataset}')
print('-----------------------')
print(f'Number of graphs: {len(dataset)}')
print(f'Number of nodes: {dataset[0].x.shape[0]}')
print(f'Number of features: {dataset.num_features}')
print(f'Number of classes: {dataset.num_classes}')
在数据集中,输入特征向量是三维的,用独热编码的形式表示,输出为0或者1表示是否为酶。
2、预处理数据集
为了利用Torch库,我们需要对数据集进行预处理。
# Create training, validation, and test sets
train_dataset = dataset[:int(len(dataset)*0.8)]
val_dataset = dataset[int(len(dataset)*0.8):int(len(dataset)*0.9)]
test_dataset = dataset[int(len(dataset)*0.9):]
print(f'Training set = {len(train_dataset)} graphs')
print(f'Validation set = {len(val_dataset)} graphs')
print(f'Test set = {len(test_dataset)} graphs')
# Create mini-batches
train_loader = DataLoader(train_dataset, batch_size=64, shuffle=True)
val_loader = DataLoader(val_dataset, batch_size=64, shuffle=True)
test_loader = DataLoader(test_dataset, batch_size=64, shuffle=True)
print('\nTrain loader:')
for i, batch in enumerate(train_loader):
print(f' - Batch {i}: {batch}')
print('\nValidation loader:')
for i, batch in enumerate(val_loader):
print(f' - Batch {i}: {batch}')
print('\nTest loader:')
for i, batch in enumerate(test_loader):
print(f' - Batch {i}: {batch}')
首先将1113个蛋白质图划分成训练集、验证集和测试集。其次,将训练集、验证集和测试集中的所有蛋白质图以64个为一个批次,组成若干个批次,用于后续的模型训练。
得到输出结果如下:
Training set = 890 graphs
Validation set = 111 graphs
Test set = 112 graphs
Train loader:
- Batch 0: DataBatch(edge_index=[2, 7646], x=[2070, 3], y=[64], batch=[2070], ptr=[65])
- Batch 1: DataBatch(edge_index=[2, 10936], x=[2984, 3], y=[64], batch=[2984], ptr=[65])
- Batch 2: DataBatch(edge_index=[2, 9092], x=[2511, 3], y=[64], batch=[2511], ptr=[65])
- Batch 3: DataBatch(edge_index=[2, 8874], x=[2510, 3], y=[64], batch=[2510], ptr=[65])
- Batch 4: DataBatch(edge_index=[2, 13412], x=[3378, 3], y=[64], batch=[3378], ptr=[65])
- Batch 5: DataBatch(edge_index=[2, 8520], x=[2234, 3], y=[64], batch=[2234], ptr=[65])
- Batch 6: DataBatch(edge_index=[2, 8466], x=[2207, 3], y=[64], batch=[2207], ptr=[65])
- Batch 7: DataBatch(edge_index=[2, 9434], x=[2641, 3], y=[64], batch=[2641], ptr=[65])
- Batch 8: DataBatch(edge_index=[2, 11142], x=[2983, 3], y=[64], batch=[2983], ptr=[65])
- Batch 9: DataBatch(edge_index=[2, 8654], x=[2359, 3], y=[64], batch=[2359], ptr=[65])
- Batch 10: DataBatch(edge_index=[2, 7162], x=[1905, 3], y=[64], batch=[1905], ptr=[65])
- Batch 11: DataBatch(edge_index=[2, 9204], x=[2525, 3], y=[64], batch=[2525], ptr=[65])
- Batch 12: DataBatch(edge_index=[2, 7664], x=[2028, 3], y=[64], batch=[2028], ptr=[65])
- Batch 13: DataBatch(edge_index=[2, 6964], x=[1830, 3], y=[58], batch=[1830], ptr=[59])
Validation loader:
- Batch 0: DataBatch(edge_index=[2, 10650], x=[2774, 3], y=[64], batch=[2774], ptr=[65])
- Batch 1: DataBatch(edge_index=[2, 5418], x=[1473, 3], y=[47], batch=[1473], ptr=[48])
Test loader:
- Batch 0: DataBatch(edge_index=[2, 11258], x=[3039, 3], y=[64], batch=[3039], ptr=[65])
- Batch 1: DataBatch(edge_index=[2, 7592], x=[2020, 3], y=[48], batch=[2020], ptr=[49])
我们以训练集的Batch 0为例,说明处理后的结果。
首先可以观察到将64个蛋白质图组合一个批次,可以得到64个 y y y值,也就是64个输出结果,判断是否为酶。其次,每一个蛋白质图中都有若干个节点,每一个节点都有一个特征向量,所有的特征向量组合成了一个 x x x,在Batch 0中,有2070个节点,每个节点的特征向量都为3。 e d g e _ i n d e x edge\_index edge_index中存储了两个相连节点的索引,第一行为起始节点的索引,第二行为目标节点的索引,在Batch 0中一共有7646个边。 B a t c h Batch Batch中存储了节点所归属的蛋白质图,例如索引为0-10的节点归属于蛋白质0,则其索引对应的数值为0。
3、类函数定义
class GCN(torch.nn.Module):
"""GCN"""
def __init__(self, dim_h):
super(GCN, self).__init__()
self.conv1 = GCNConv(dataset.num_node_features, dim_h)
self.conv2 = GCNConv(dim_h, dim_h)
self.conv3 = GCNConv(dim_h, dim_h)
self.lin = Linear(dim_h, dataset.num_classes)
def forward(self, x, edge_index, batch):
# Node embeddings
h = self.conv1(x, edge_index)
h = h.relu()
h = self.conv2(h, edge_index)
h = h.relu()
h = self.conv3(h, edge_index)
# Graph-level readout
hG = global_mean_pool(h, batch)
# Classifier
h = F.dropout(hG, p=0.5, training=self.training)
h = self.lin(h)
return F.log_softmax(h, dim=1)
class GIN(torch.nn.Module):
"""GIN"""
def __init__(self, dim_h):
super(GIN, self).__init__()
self.conv1 = GINConv(
Sequential(Linear(dataset.num_node_features, dim_h),
BatchNorm1d(dim_h), ReLU(),
Linear(dim_h, dim_h), ReLU()))
self.conv2 = GINConv(
Sequential(Linear(dim_h, dim_h),
BatchNorm1d(dim_h), ReLU(),
Linear(dim_h, dim_h), ReLU()))
self.conv3 = GINConv(
Sequential(Linear(dim_h, dim_h),
BatchNorm1d(dim_h), ReLU(),
Linear(dim_h, dim_h), ReLU()))
self.lin1 = Linear(dim_h * 3, dim_h * 3)
self.lin2 = Linear(dim_h * 3, dataset.num_classes)
def forward(self, x, edge_index, batch):
# Node embeddings
# WL algorithm: Consider adjacency nodes several times
h1 = self.conv1(x, edge_index)
h2 = self.conv2(h1, edge_index)
h3 = self.conv3(h2, edge_index)
# Graph-level readout
# sum global pooling
h1 = global_add_pool(h1, batch)
h2 = global_add_pool(h2, batch)
h3 = global_add_pool(h3, batch)
# Concatenate graph embeddings
h = torch.cat((h1, h2, h3), dim=1)
# Classifier
h = self.lin1(h)
h = h.relu()
h = F.dropout(h, p=0.5, training=self.training)
h = self.lin2(h)
return F.log_softmax(h, dim=1)
def train(model, loader):
criterion = torch.nn.CrossEntropyLoss()
optimizer = torch.optim.Adam(model.parameters(), lr=0.01)
epochs = 100
model.train()
for epoch in range(epochs + 1):
total_loss = 0
acc = 0
val_loss = 0
val_acc = 0
# Train on batches
for data in loader:
optimizer.zero_grad()
batch_num_counter = Counter(data.batch.numpy())
out = model(data.x, data.edge_index, data.batch)
loss = criterion(out, data.y)
total_loss += loss / len(loader)
acc += accuracy(out.argmax(dim=1), data.y) / len(loader)
loss.backward()
optimizer.step()
# Validation
val_loss, val_acc = test(model, val_loader)
# Print metrics every 20 epochs
if (epoch % 20 == 0):
print(
f'Epoch {epoch:>3} | Train Loss: {total_loss:.2f} | Train Acc: {acc * 100:>5.2f}% | Val Loss: {val_loss:.2f} | Val Acc: {val_acc * 100:.2f}%')
return model
@torch.no_grad()
def test(model, loader):
criterion = torch.nn.CrossEntropyLoss()
model.eval()
loss = 0
acc = 0
for data in loader:
out = model(data.x, data.edge_index, data.batch)
loss += criterion(out, data.y) / len(loader)
acc += accuracy(out.argmax(dim=1), data.y) / len(loader)
return loss, acc
def accuracy(pred_y, y):
"""Calculate accuracy."""
return ((pred_y == y).sum() / len(y)).item()
在上述代码中, G C N GCN GCN类就不介绍了,需要了解可查看GCN。着重介绍一下本文的主体内容GIN类。
在GIN类中,首先定义了三个 G I N C o n v GINConv GINConv函数,在该函数中有线性变换、归一化处理和激活函数处理。这一部分主要对应于WL检验部分,多次考虑邻接节点。邻接节点经过聚合和组合的操作后,得到相对应的嵌入向量。其次,定义了 g l o b a l _ a d d _ p o o l global\_add\_pool global_add_pool池化函数,在默认参数下,主要对嵌入向量进行累加。最后,通过 c a t cat cat将蛋白质在不同层的嵌入向量相连接经过 M L P MLP MLP后输出是否是酶的概率。
注意到 G I N C o n v GINConv GINConv需要输入 e d g e _ i n d e x edge\_index edge_index,意味着是对邻接节点进行相应操作。而在 g l o b a l _ a d d _ p o o l global\_add\_pool global_add_pool中需要输入 b a t c h batch batch,也就是说需要判断节点是否属于同一个蛋白质,并将同一个蛋白质的节点嵌入向量求和得到结果。
4、模型训练
gin = GIN(dim_h=32)
gin = train(gin, train_loader)
test_loss, test_acc = test(gin, test_loader)
print(f'Test Loss: {test_loss:.2f} | Test Acc: {test_acc * 100:.2f}%')
print()
gcn = GCN(dim_h=32)
gcn = train(gcn, train_loader)
test_loss, test_acc = test(gcn, test_loader)
print(f'Test Loss: {test_loss:.2f} | Test Acc: {test_acc*100:.2f}%')
fig, ax = plt.subplots(4, 4)
fig.suptitle('GCN - Graph classification')
for i, data in enumerate(dataset[-48:-32]):
# Calculate color (green if correct, red otherwise)
out = gcn(data.x, data.edge_index, data.batch)
color = "green" if out.argmax(dim=1) == data.y else "red"
# Plot graph
ix = np.unravel_index(i, ax.shape)
ax[ix].axis('off')
G = to_networkx(dataset[i], to_undirected=True)
nx.draw_networkx(G,
pos=nx.spring_layout(G, seed=0),
with_labels=False,
node_size=10,
node_color=color,
width=0.8,
ax=ax[ix]
)
plt.show()
fig, ax = plt.subplots(4, 4)
fig.suptitle('GIN - Graph classification')
for i, data in enumerate(dataset[-48:-32]):
# Calculate color (green if correct, red otherwise)
out = gin(data.x, data.edge_index, data.batch)
color = "green" if out.argmax(dim=1) == data.y else "red"
# Plot graph
ix = np.unravel_index(i, ax.shape)
ax[ix].axis('off')
G = to_networkx(dataset[i], to_undirected=True)
nx.draw_networkx(G,
pos=nx.spring_layout(G, seed=0),
with_labels=False,
node_size=10,
node_color=color,
width=0.8,
ax=ax[ix]
)
plt.show()
训练 G C N GCN GCN和 G I N GIN GIN模型,并在测试集上检验模型预测的准确率,最后可视化预测结果。
GCN accuracy: 73.96%
GIN accuracy: 77.86%
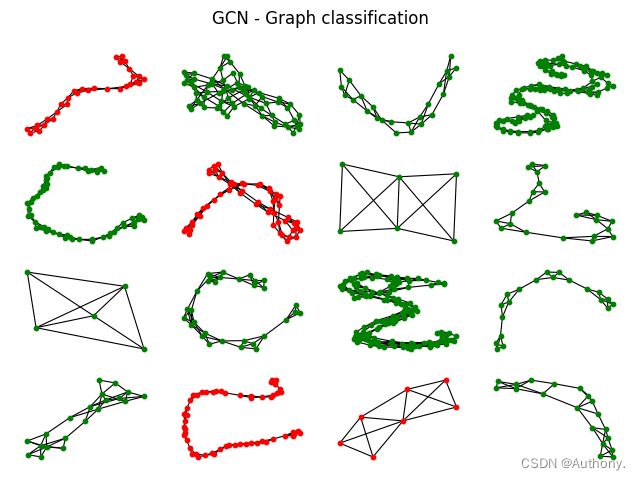
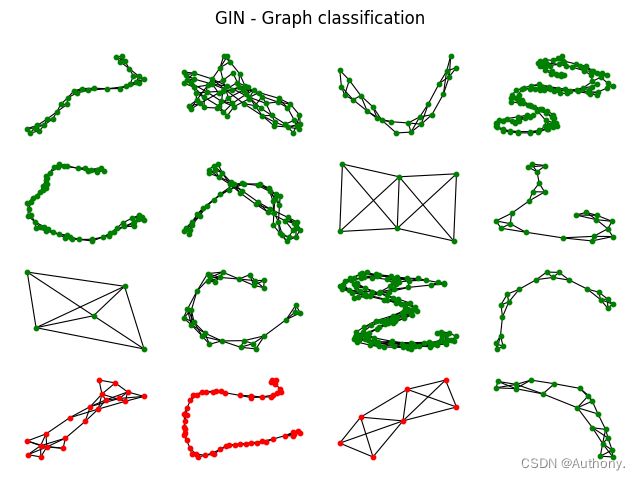
在图4中可以发现, G C N GCN GCN和 G I N GIN GIN模型在错误结果的预测上具有一定的相似性,我们可以考虑重新选择能够在 G C N GCN GCN和 G I N GIN GIN预测错误的结果上表现较好的模型,之后利用结合三者的预测结果。这可能提高预测的准确率。
总结
在本章中,我们提出了WL检验方法,它可以输出图的规范形式。该算法并不完美,但可以区分大多数图结构。它启发了GIN体系结构,设计成与WL测试一样具有表现力,因此严格来说比GCNs、GATs或GraphSAGE更具表现力。然后我们实现了这个架构用于图分类。我们看到了将节点嵌入组合成图嵌入的不同方法。GIN提供了一种新的技术,它结合了求和算子和每一层生成的图嵌入的连接。它明显优于GCN层获得的经典全局平均池化。最后,我们比较了两种模型的预测结果,并提出了进一步提高模型预测结果的可能方法。