用于解释非目标代谢组学数据的集成深度学习框架
摘要
非定向代谢组学正获得广泛应用。数据分析的关键方面包括建模代谢网络的复杂活动、选择与临床结果相关的代谢物以及发现关键代谢途径以揭示生物学机制。数据分析中的一个关键障碍未得到很好解决,即数据特征与已知代谢物之间的匹配不确定性问题。鉴于实验技术的限制,数据特征的身份不能直接在数据中揭示。
将特征映射到代谢物的主要方法是将数据特征的质荷比(m/z)与已知代谢物的理论值匹配。由于一些代谢物共享分子组成,同一代谢物可以衍生出不同的附加物离子,特征与代谢物之间的关系并非一一对应。这种匹配不确定性导致了不可靠的代谢物选择和功能分析结果。
在这里,我们介绍了一种考虑匹配不确定性的集成深度学习框架,用于代谢组学数据。该模型设计有一个基于已知代谢网络和特征与代谢物之间注释关系的渐进稀疏化神经网络。该体系结构刻画了代谢组学数据并反映了生物系统的模块化结构。三个目标可以同时实现,而无需进行复杂的推断和额外的假设:
(1)评估代谢物的重要性
(2)推断特征-代谢物匹配的可能性
(3)选择疾病亚网络。当应用于COVID代谢组学数据集和老龄化小鼠脑数据集时,我们的方法找到了容易解释的代谢亚网络。
介绍
近年来,越来越多的研究试图探索代谢组学,以揭示疾病病理,寻找早期干预策略[1]。非靶向代谢组学数据是通过对生物样品中小分子物质的无偏测量而获得的,能够反映生物体内调控和代谢途径的功能变化。当分析疾病样本时,代谢组直接反映了身体的病理状态,从而导致了对其他形式的组学测量的补充发现[2,3]。
目前,收集非靶向代谢组学数据的主要方法是使用液相色谱-质谱(LC/MS)。LC/MS 数据具有很高的噪音,并且预处理包括多个步骤,如峰检测、对齐、保留时间校正、弱信号恢复等[4–6]。在LC/MS数据预处理之后,每个特征都通过质荷比(m/z)、保留时间(RT)和样本中的强度进行表征。为了确定特征的分子身份,通常的方法是基于质荷比(m/z)将特征与已知代谢物进行匹配。由于一些代谢物共享分子组成,同一代谢物可以衍生出不同的附加物离子,一个特征可以与多个已知代谢物匹配,一个代谢物也可以与多个特征匹配[7]。最近对数据无关采集(DIA)的进展使得代谢物注释更加准确。然而,当前阶段DIA仍不太适用于大规模研究[8]。已经开发了几种用于LC/MS数据的方法,通过使用已知代谢物作为参考,这些方法融入了代谢物之间的反应相似性[9]。
"数据无关采集"(Data-Independent Acquisition,DIA)是一种质谱分析方法,它的目标是获取样本中所有离子的质谱数据,而不是仅仅选择特定的离子进行分析。在DIA中,质谱仪会按照一定的质荷比窗口(m/z窗口)连续地扫描整个质谱范围,而不是选择性地监测特定的质荷比。这种方法相对于数据相关采集(Data-Dependent Acquisition,DDA)来说,更全面地捕获了样本中的信息。
LC/MS代谢组学数据分析涉及三个主要任务。
- 首先是选择与研究的临床结果相关的特征。
- 其次是找到与显著特征相对应的代谢物。
- 第三是确定哪些代谢途径受到生物条件的影响。
通常与特定临床结果相关的代谢物只占所有代谢物的一小部分。确定有效的代谢物对于理解潜在的生物学机制至关重要。存在许多用于高维特征选择的方法[10, 11]。然而,鉴于代谢组学数据中的匹配不确定性,所选特征与代谢物之间并没有一对一的对应关系,这使得难以明确确定重要的代谢物。
除了代谢物的选择之外,代谢网络或途径分析是代谢组学数据分析的关键部分[12]。整合网络知识使生物标志物签名的发现更加稳健、稳定和可解释[13]。已经开发了许多用于使用基因表达数据进行子网络选择的方法[14–16]。同样,在代谢组学数据分析中,特征-代谢物匹配引入了额外的不确定性,这是上述方法无法解决的。
在专门为代谢组学数据开发的一些方法中,一些途径分析方法忽略了匹配不确定性问题[17, 18]。考虑到每个特征只能有一个正确的匹配,一些最近的研究尝试通过对匹配进行统计推断来消除/减少不确定性。特征之间的关系,比如符合常见附加物离子理论差异的质荷比差异,以及相似的保留时间,可以帮助确定两个特征是否可能来自同一代谢物[7, 19]。Cai等人[20]尝试将最佳匹配选择与预测模型中的特征选择相结合,但选择是二元的且缺乏适当的推断。Shen等人[21]利用了MS2的相似性,这通常对于大多数特征不可用,在反应对配邻域中推断真实匹配。这些工作并未提供代谢组学数据分析的集成流程,因为它们没有系统地计算潜在特征-代谢物匹配的可能性,并且它们没有评估个体代谢物对预测的重要性,这两者都是下游分析的基础。此外,当前方法不允许在代谢物丰度和疾病状态之间进行灵活建模,而这经常涉及非线性关系。为了填补这一空白,我们提出了一个统一框架,同时实现三个目标:(1)评估代谢物重要性,(2)推断特征-代谢物匹配的可能性,以及(3)从整体代谢网络中选择疾病亚网络。
为实现这一目标,我们采用了深度神经网络方法,该方法在许多组学领域取得了良好的性能[22–25]。我们还从最近关于通过添加特定结构或损失函数将领域知识融入神经网络的研究中汲取了灵感[26]。我们设计了一种新颖的深度学习模型,该模型包含一个基于特征-代谢物匹配和已知代谢网络的逐渐稀疏化结构,并设计了中间变量重要性和边缘重要性的新度量,以找到重要的代谢物和最可能的特征-代谢物匹配。在技术上,该方法可以被视为基于知识图的结构稀疏模型,因为它包含一个逐层逐渐稀疏化的结构,以更好地反映生物系统的模块化特性。在应用方面,我们新提出的方法可以作为分析非定向代谢数据的便利工具。其稀疏结构避免了在高维数据 - 低样本量(N << p)情境中的过度参数化,并倾向于选择落入亚网络的代谢物。它实现了良好的代谢物选择结果,并同时推断了最可能的特征-代谢物匹配。
3. **倾向于选择落入亚网络的代谢物:**
- "亚网络"指的是整体代谢网络中的子网络或相关联的集合。
- 由于采用了稀疏结构,该方法更有可能选择与生物学相关性更强、更有意义的代谢物,这些代谢物可能在生物学系统中形成特定的亚网络。因此,该方法通过在高维数据和低样本量情境中采用稀疏结构,有助于提高模型的泛化能力,选择更具生物学意义的代谢物,并更好地反映生物系统的模块化结构。
方法
方法概述
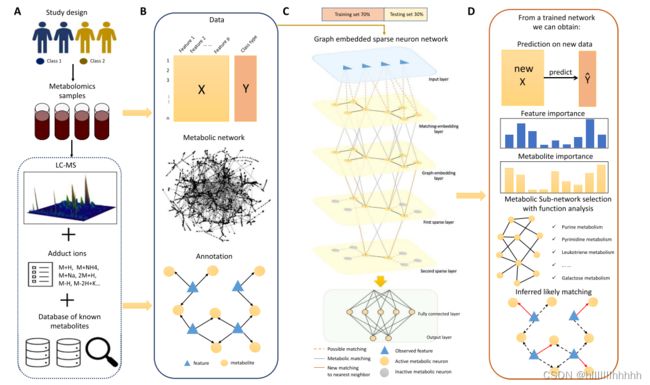
我们提出了一个用于分析非定向代谢组学数据的统一深度学习框架(图1)。该框架利用特征-代谢物注释关系和已知代谢网络来构建逐层逐渐稀疏的神经网络。该模型以全面的方式分析非定向代谢组学数据,支持分类、代谢物/亚网络选择以及推断特征与代谢物之间可能的匹配等任务。
该方法以一个特征丰度矩阵和一个包括潜在特征与代谢物匹配关系的表格作为起点,这个表格可以通过诸如xMSAnnotator等工具获得[27]。众所周知,存在一个匹配不确定性问题。由于许多代谢物共享相同的分子组成,因此一个特征可以与多个代谢物匹配。同时,每个代谢物可以生成多个附加物离子(图1A)。我们提出的框架将两种现有连接嵌入到一个稀疏神经网络中,这两种连接分别是:
(1)特征-代谢物潜在匹配和
(2)通过代谢网络中的共同反应的代谢物连接(图1B)。
总体来说,这个框架通过将这两种连接嵌套到神经网络中,以更好地考虑特征与代谢物之间的潜在匹配关系,同时也综合了代谢网络中的信息,使得模型能够更全面地分析代谢组学数据。这有助于解决匹配不确定性问题,提高分析的鲁棒性和可解释性。
在神经网络中,第一个隐藏层被命名为匹配嵌入层。它的隐藏神经元与代谢物一一对应。输入节点与这些隐藏神经元之间的连接由特征与代谢物之间的注释关系确定(图1C)。在第二个隐藏层中,我们将代谢网络结构嵌入为图嵌入层。该层再次包含一一对应于代谢物的神经元。
图嵌入层中的神经元仅在已知代谢网络中通过反应连接的相应代谢物对之间建立连接。在图嵌入层之后是几个逐渐稀疏化的层。这些层中每一层只包含在已知代谢网络中具有连接度 ≥ 预先指定阈值的神经元。层次越深,阈值越高。在达到足够的稀疏性后,接下来是传统的全连接层和输出层(图1C)。
整体而言,这个设计意味着通过逐渐稀疏化的处理,模型更集中地关注具有较高连接度的代谢物,以更有效地捕捉代谢网络中的关键信息。
该网络通过使用Adam优化器最小化交叉熵损失在训练数据集上进行训练。基于训练好的模型,我们可以实现四个目标:(1)对新数据进行预测,(2)评估代谢物和特征的重要性,(3)进行代谢物和亚网络的选择进行功能分析,以及(4)推断可能的特征-代谢物匹配(图1D)。
图1. 综合深度学习框架概述。
- (A) 数据来源的图示,包括分析LC-MS数据以生成数据特征(样本间对齐的峰值),以及将特征映射到已知代谢物。
- 输入代谢组学样本——LC-MS进行分析——加合物图标——已知代谢物数据库
- (B) 综合信息,包括特征丰度矩阵与临床结果、已知代谢网络结构和潜在特征匹配到代谢物的信息。
- 特征丰度矩阵——代谢网络——匹配特征信息
- (C) 逐层逐渐稀疏的神经网络,以特征表达数据为输入,样本类别为输出。它包括三个部分:基于潜在特征注释的特征-代谢物嵌入、代谢网络嵌入和逐层逐渐稀疏化、以及全连接层。
- 输入层——匹配嵌入层——图嵌入层——第一稀疏层——第二稀疏层——全连接层——输出层
- (D) 经过训练的模型的结果。该模型可以进行分类,确定可能的特征-代谢物匹配,进行代谢物和代谢子网络的选择,用于功能分析。
基于知识图的稀疏神经网络模型
我们新提出的模型是一个逐层逐渐稀疏的神经网络,它逐渐聚集在对应于输入图中枢节点的神经元周围的信号。这种稀疏结构的设计旨在解决训练数据有限和当网络随着大量输入变量变宽时出现的计算负载急剧增加的问题[28]。当没有输入变量之间的知识图可用时,获得稀疏网络的一般方法是从训练一个全连接网络开始,然后迭代地修剪连接。已经验证了适当的稀疏网络在准确性上可以与全连接网络相媲美[29]。
在组学数据的情况下,要考虑的变量数量通常在数千数量级,而样本大小通常在数百个。存在一个描述变量之间功能关系的知识图。利用知识图在两个方面是有益的:
(1)在样本量较小时,实现更具鲁棒性的稀疏模型训练
(2)产生符合现有知识的变量选择结果,使结果更具可解释性。
由于特征-代谢物匹配的不确定性,非定向代谢组学数据尤为具有挑战性。代谢网络可用于描述代谢物之间的功能关系,我们的兴趣是找出与临床结果最相关的代谢物和代谢途径(整个代谢网络的子网络)。
我们的模型建立在以下假设的基础上。
- 第一假设是每个特征在所有给定的潜在匹配中都有其真实的注释。
- 第二假设是只有很小一部分的代谢物对疾病结果有真实的预测作用,并且它们倾向于位于完整代谢网络的小子网络中。这些假设在先前的研究中被广泛使用和承认。
为了从原始特征丰度数据中找到有效的代谢物,我们的模型通过在统一框架中将潜在的匹配关系与代谢网络结合起来,解决了匹配不确定性的问题。

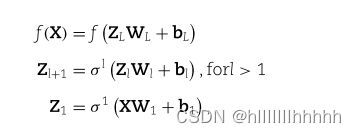





输入层与第一隐层(匹配嵌入层)之间的连接由矩阵M确定
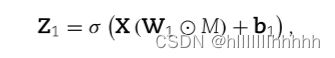

使用逐元素乘法是为了确定原始矩阵中哪些元素重要
在下一个稀疏层中,我们首先确定一个稀疏化因子 μ(0 < μ < 1),然后决定稀疏层的大小为 |L3| = m × μ。根据这个数量,我们包括在 G 中度排名最高的节点,这些节点被称为活跃节点。我们将未被选择进入下一个稀疏层的节点称为非活跃节点。活跃节点的连接从代谢网络继承而来。一些非活跃节点没有连接到下一层,因为它们在代谢网络上的第一邻居都是非活跃节点。对于这样的节点,通过将非活跃节点链接到其在代谢网络上的最近邻的活跃节点之一来添加新连接。

类似地,我们构建了几个稀疏连接层,超过第 3 个隐藏层,每个都比前一个小。在稀疏神经网络部分之后,我们附加全连接层,最后一层输出不同类别的概率预测。
特征和代谢物重要性评价
识别预测性代谢物对于下游分析至关重要,有助于揭示潜在的生物学机制,从而更好地理解临床结果。我们从训练好的模型中推断真实的匹配,并评估特征和代谢物的重要性。这个想法类似于 [23] 引入的图连接权重(Graph Connection Weights,GCWs)方法。在GCW中,一个预测变量的重要性由其相关权重的大小反映。我们的提议是,在反向传播训练过程中,真实匹配的权重在过程中获得更多的关注。
真实匹配的权重在过程中获得更多的关注。
意思是说能够找到匹配的代谢物
首先,我们考虑代谢物重要性的估计。与 GCW 相比,我们尝试消除链接数量的影响,其中包括与特征的匹配数量,以及在代谢网络中的度。为了避免零分母,我们对每个代谢物的代谢物-特征链接数添加了1。对于第一和第二隐藏层的神经元,每个神经元与一个代谢物有一对一的映射,因此对其关联权重的绝对值求和得到了一个重要性的估计,即:
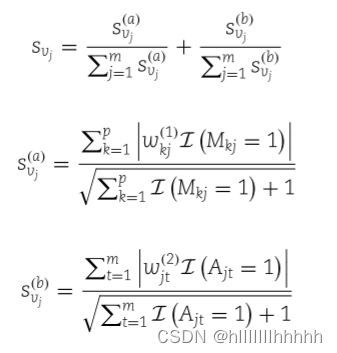

类似地,我们可以使用第一层中的权重来推断特征的重要性。对于每个特征,我们将其重要性定义为其潜在相关代谢物的重要性之和,即



详细的模型设置

结果
COVID代谢组学数据分析
在前所未有的全球冠状病毒病 2019(COVID-19)大流行期间,代谢组学技术已被采用来研究 COVID 感染的代谢反应。理解代谢模式与疾病严重程度之间的关联,并识别可能导致严重疾病结果的生理过程至关重要。Metabolomics Workbench 上的 ST001849 数据集是为了找到 COVID 感染的预后标志物而收集的[32]。我们将我们的模型应用于在患者入院时(第0天)收集的血浆代谢组学数据的子集,以找到与患者后来是否被送入重症监护室(ICU)相关的代谢物和代谢途径。
我们下载了原始的液相色谱/质谱(LC/MS)数据,并使用 apLCMS [6, 33] 进行了数据预处理,随后使用 combat [34] 进一步处理以消除批次效应。我们移除了在超过 75% 样本中具有零丰度的特征,并进行了 log(1+x) 转换。对数据进行了最小-最大归一化和等距投影,以统一数据的尺度。使用 xMSannotator [27] 进行了特征到代谢物的注释。分析使用了KEGG代谢网络[35]。在过滤后,我们得到了一个包含 1351 个特征、匹配到 913 个代谢物的数据集,共包括 263 个样本,其中 123 个样本后来被送入了重症监护室(标签 1),而 140 个样本未被送入(标签 0)(图2A)。每个代谢物的平均匹配值为3.11,范围为1-13。在观察的特征方面,平均匹配值为2.11。
该模型在两个稀疏层和一个具有20个隐藏神经元的全连接层中使用了0.3的稀疏率。全连接层中的丢失率为0.3。百分之七十的样本被选为训练集,其余的用作测试集。我们使用批量大小为32,在Adam优化器下对模型进行了100个epochs的训练。进行了十次实验,最终代谢物的重要性分数是在这十次实验中取平均得到的。平均测试准确率为0.765。该方法为每个代谢物输出一个重要性分数。为了选择重要的代谢物,我们使用了在AIME [36]中提出的具有伽马零分布的局部假发现率(lfdr)程序。我们选择了113个代谢物,其估计的lfdr ≤ 0.1。
96个重要代谢物中的大多数都位于图2B所示的一个相对紧密的亚网络中。为了识别与其他患者相比被送入重症监护室的患者中发生代谢途径失调,我们进一步进行了基于超几何测试的代谢途径富集分析,使用了'metapone'软件包中嵌入的代谢途径数据库[37]。选定代谢途径的一些显著代谢物显示在图3中。我们观察到中心代谢,包括能量、氨基酸和核酸代谢,都与疾病的严重程度相关。
例如,14个显著代谢物属于天冬氨酸和天门冬氨酸代谢,22个属于酪氨酸代谢,8个属于尿素循环,7个属于精氨酸和脯氨酸代谢。氨基酸代谢的其他变化包括苯丙氨酸代谢等。已经发现在尿素循环中,重症患者的鸟氨酸浓度较高。相比之下,COVID-19患者中精氨酸浓度显著降低,精氨酸向鸟氨酸的转化在尿素循环中占主导地位[38]。据信,精氨酸的降解加剧了炎症反应。它导致其下游代谢物(如鸟氨酸)的积累。这些变化最终限制了天冬氨酸的消耗,从而增加了天冬氨酸的浓度。这一系列效应已被证明影响T细胞激活水平[39, 40]。与此同时,天冬酸及其下游产物天门冬酰胺的增加为病毒mRNA的翻译提供了有利的环境。因此,在COVID-19的发展过程中,机体失去了调节精氨酸的能力,导致尿素循环失衡和更严重的炎症损伤。苯丙氨酸代谢在COVID患者的重症和死亡组中富集[41],可能是由于严重的炎症反应消耗了大量四氢生物蝶啶,从而限制了肝细胞中苯丙氨酸向酪氨酸的转化。与此同时,升高的血清苯丙氨酸进一步加剧了炎症过程[42]。
嘧啶和嘌呤代谢也是排名靠前的途径。已有研究报告称,胞嘧啶是重症急性呼吸综合症冠状病毒2型(SARS-CoV-2)细胞代谢的协调物,是病毒演变的关键[43]。关于炎症反应的研究表明,抑制嘧啶生物合成可以减少炎症细胞因子的释放,而抑制嘌呤代谢会增强炎症反应[44, 45]。免疫反应失调可能导致细胞因子风暴,引发严重的肺部炎症,甚至引发全身性连锁反应,如脓毒性休克和多器官功能衰竭[46]。在ICU的患者中,IL-2、IL-6、IL-7、IL-10和巨噬细胞炎症蛋白的表达水平较高[47]。尼克酰胺导致多ADP核糖聚合酶(PARP)活性的抑制,从而减少一氧化氮合酶的表达。这一过程减少了自由基和促炎细胞因子[48]。因此,人们认为通过补充尼克酸和尼克酰胺可以减少促炎细胞因子。这解释了我们发现的尼克酸和尼克酰胺代谢途径。
其他显著的途径包括碳代谢、萜类物质和类固醇的生物合成、三羧酸循环和丁酸代谢。研究表明,DNA 和 RNA 病毒通过改变中心碳代谢途径,包括糖酵解、三羧酸循环、氨基酸合成/降解等,重构寄主细胞的新陈代谢[49]。由于能量产生大幅减少和细胞能量代谢紊乱,COVID-19 患者体内葡萄糖和丙酮酸水平升高[50]。同时,由于肝功能异常变化,导致胃肠道通透性增加,氨基酸和蛋白质的消化和吸收异常[51]。在比较重症和轻症 COVID-19 患者时,发现 COVID-19 的发展导致体内乳酸增加,可能是由于疾病进展中氧气吸入和营养摄入不足[38]。
总体而言,我们的方法从 COVID 数据集中选择了合理的代谢物和代谢途径。同时,它能够为特征同时分配重要性得分。在图3中,黄色节点表示数据特征。颜色越深,重要性得分越高。该方法还为潜在的特征-代谢物匹配分配了置信水平(图3中的灰色边缘)。较粗的灰色边缘表示匹配的置信度更高。
Urea cycle 尿素循环
Tyrosine metabolism 酪氨酸代谢
Arginine and proline metabolism 精氨酸与脯氨酸代谢
Phenylalanine metabolism 苯丙氨酸代谢
Aspartate and asparagine metabolism 天冬氨酸和天冬酰胺代谢
Butanoate metabolism 丁酸代谢
Carbon metabolism 碳代谢
Tca cycle TCA循环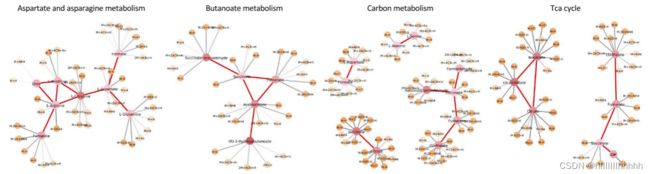
Purine metabolism 嘌呤代谢
Pyrimidine metabolism 嘧啶代谢
Nicotinate and nicotinamide metabolism 烟酸与烟酰胺代谢
Biosynthesis of terpenoids and steroids 萜类化合物和甾体的生物合成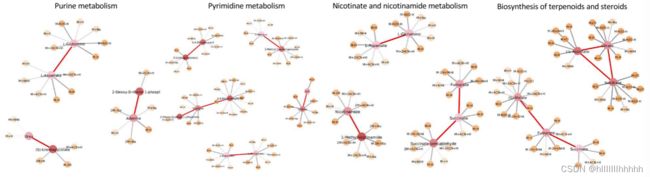
图3。与COVID病情严重程度相关的亚网络。选择的代谢亚网络,其中代谢物节点较大,特征节点较小。节点的阴影表示代谢物/特征的重要性。颜色越深,重要性得分越高。特征和代谢物之间边缘的粗细表示基于模型的边缘置信度。
衰老小鼠脑数据的应用
我们在代谢组学工作台的老年小鼠脑数据上应用了我们的方法[52]。在哺乳动物中,大脑在老化过程中发生显著变化,导致认知下降、记忆丧失和睡眠模式改变。对大脑在老化过程中代谢变化的全面理解尚不足。我们尝试利用老年小鼠脑代谢组学数据揭示健康老化过程中的变化。
小鼠脑数据集包含16只小鼠(8只雄性和8只雌性),分为三个年龄阶段:AD(3周),EA(16周,早期成年)和MA(59周,中年),采集于大脑的10个区域。这使得总样本量为480。我们删除了在样本中75%具有零丰度的特征,并选择了与KEGG代谢网络[35]上的代谢物匹配的特征。最终,我们得到了一个包含671个特征匹配到713个代谢物的数据集。我们进行了对数变换,并使用最小-最大规范化将数据缩放到-500到500的范围内。我们使用在EA和MA时期采集的样本来拟合我们的模型,以研究健康老化对大脑的影响,涉及320个样本。分类任务是区分MA样本和EA样本。我们将所有十个脑区的数据合并,以找到大脑健康老化的一般代谢变化模式。为了整合脑区信息,我们添加了10个神经元,其中脑区由one-hot编码表示,并连接到第一个全连接层。
由于数据来自10个脑区,其多样性增加了训练的难度。因此,我们在每个层的修正线性单元(ReLU)激活之后添加了一个批量归一化层,以稳定训练过程。使用70%的样本进行训练,其余用作测试集。使用Adam优化器,我们以64的批量大小训练了200个时代的模型。代谢物和特征的重要性分数,以及代谢物-特征匹配的置信度,在十次重复实验中进行了平均。测试集上的平均分类准确性为0.8750。使用具有伽马零分布的局部fdr程序,估计的lfdr ≤ 0.05的代谢物被视为显著。选择了56个代谢物。图4显示了选择的代谢子网络及其功能注释。
衰老导致突触损伤和神经系统功能障碍。现有研究指出,在老化过程中海马发生神经生物学变化,如氧化应激增加、神经炎症和细胞内信号传导的改变 [53]。这一系列变化可能导致神经退行性疾病的发生,如阿尔茨海默病和失忆症。在选择的代谢子网络中,最大的组分涉及糖代谢,第二大数量的子网络主要与神经信号传导有关。
改变的神经信号传导是衰老的标志。我们的方法发现了两条显著的通路,包括“5-羟色胺能突触”和“炎性介质调控色氨酸通道”。5-羟色胺(5-HT)是一种调节许多生理目标的单胺神经递质。Saleem等人 [54] 发现老年大鼠的额叶皮质和海马中检测到5-HT水平下降。同时,还发现阿尔茨海默病和失忆症患者的5-HT也减少 [55],这表明5-HT参与了与神经元相关的认知衰退。与5-羟色胺能突触途径部分重叠的TRP通道途径在调节与各种细胞功能相关的钙平衡方面起着关键作用 [56]。TRP通道的异常可能导致过多的钙流入,破坏pH水平和神经元稳态 [57],从而导致与年龄相关的神经退行性疾病。
大脑衰老过程也与身体内炎症的增加密切相关。包括“半乳糖代谢”、“亚油酸代谢”、“花生四烯酸代谢”和“白三烯代谢”等通路参与了炎症过程。已经显示半乳糖代谢能够诱导颗粒回旋回(DG)中神经祖细胞中反应性氧化物质(ROS)的积累,该过程与DG区域神经发生的减少相关 [58]。此外,亚油酸代谢产物花生四烯酸的浓度与大脑抗氧化系统中的催化酶活性相关 [59]。补充花生四烯酸有助于维持神经元膜的流动性,并保持海马可塑性,从而有益于记忆的恢复 [60]。然而,花生四烯酸及其下游产物的上调可能加剧大脑神经炎症和兴奋毒性 [61]。从老年神经纤维样斑区域,发现花生四烯酸普遍升高 [62]。作为花生四烯酸的代谢产物之一,白三烯有助于神经病理学的紊乱,并被认为是阿尔茨海默病的潜在药物靶点 [63]。
此外,受损的能量代谢还显著影响神经元活动和可塑性,这解释了我们发现的包括“嘧啶代谢”和“六碳糖磷酸化”在内的途径。许多研究发现在老化的大脑中存在细胞能量代谢的显著失衡。一项代谢研究发现老化个体中存在嘧啶的积累,另一项研究显示老化的海马神经元更有能力在糖酵解中氧化葡萄糖 [70, 71]。我们还发现“核苷酸糖代谢”和“氨基糖代谢”与大脑老化相关,这一发现与先前的研究一致 [72]。
总体而言,通过使用老年小鼠脑代谢组学数据集,我们的方法克服了匹配不确定性的问题,并能选择易于解释的代谢物和途径。结果指出了几条路径,有助于更好地理解大脑老化。我们注意到当前的研究使用了16到59周之间的健康老化数据,表明基于代谢组学数据研究大脑发育的可行性。
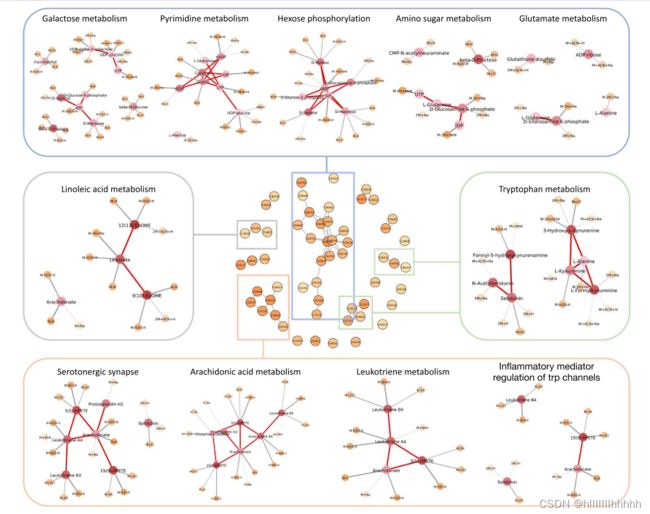
图4. 从老年小鼠脑数据中选择的代谢物。中间:选择的代谢物及其在KEGG代谢网络上的连接。子网络中局部结构的功能分析显示在不同颜色的框中。节点上颜色的深浅表示重要性得分。节点和代谢物之间的边越粗,置信度越高。
模拟研究
模拟设计
为了生成具有m个代谢物和p个特征的非靶向代谢数据,我们首先在代谢物之间生成一个无标度图。我们使用Barabási-Albert(BA)模型[73],其中幂律参数捕获了网络的度分布。在特征和代谢物之间,我们假设真实匹配是一对一映射,每个特征和代谢物的潜在匹配不超过15个。对于特征xi,我们首先随机选择其对应的真实代谢物,表示为vˆi。我们假设50%的特征有多个潜在匹配。我们从截断值的泊松分布中抽样,以确定每个特征的确切匹配数量。具体而言,对于具有多个匹配项的特征xi,可能的匹配代谢物是从距离vˆi不超过三个跳跃的代谢物池中随机选择的,且到目前为止具有不到三个匹配特征。循环遍历所有特征后,我们得到了真实匹配矩阵Mtrue和潜在匹配矩阵Mmulti。
为了在对数尺度上生成包含n个样本的特征丰度矩阵,我们使用了一个依赖于代谢物网络结构的方差-协方差矩阵的多元正态分布。从代谢网络中,我们得到了一个记录所有代谢物对的最短距离的m×m矩阵D。然后,我们确定了代谢物的协方差矩阵为(m),其中

特征之间的协方差使用协方差矩阵从其真匹配代谢物继承,
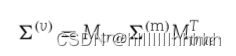
接着,使用协方差矩阵(m)的多元高斯分布来生成特征丰度矩阵X = [x1, ..., xn]T ∈ Rn×p。
为了模拟具有两个类别的输出y,我们首先选择了一些代谢物作为真正的预测因子。在假设只有代谢网络中的一小部分代谢物与结果相关,并且它们倾向于聚集在代谢网络中的派系中,我们首先随机选择一个中心代谢物(在前1%中的度)作为核心代谢物,然后向外扩展,随机包括50%的邻居顶点。这个过程迭代重复,直到真正的预测因子数量达到预设值。我们用一个二进制向量T = [t1, t2, ..., tm]T表示真正的代谢物预测因子,其中tj = 1表示代谢物vj是真正的预测因子,否则该值设为零。真正的预测特征通过S = MtrueT ∈ Rp找到。
为了模拟输出类标签y ∈ Rn,我们首先从均匀分布中采样系数参数β = (β1, β2, ..., βp)T,取值范围在[0.7, 0.9]之间,并随机将其中一些设置为负值。使用广义线性模型生成输出y,如下所示:
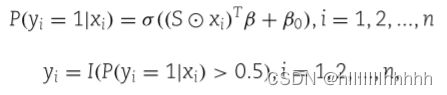
其中 ⊙ 表示逐元素乘法,Sxi 有效地将不贡献的特征设为零,σ 是逻辑连接函数,即:

按照上述步骤,我们生成了一系列具有不同设置的实验。对于所有实验,我们将特征数量设置为1000,真实代谢物预测器的比例为10%或20%。在我们的模拟研究中,我们使用了不同数量的代谢物和样本大小,即1000、2000个代谢物和2000、4000、6000和8000的样本大小。此外,我们测试了泊松分布的不同参数设置,包括 λ = 5、10 和 15,这决定了多重匹配的数量。在所有实验中,我们使用了我们提出的图嵌入稀疏神经网络来推断真实匹配,并找出潜在重要的代谢物和特征。
对于每个模拟设置,进行了10次实验。每个数据集被分为训练集和测试集,比例为7:3。模型在训练数据上进行训练,并在测试数据上进行预测。每个实验的计算时间,使用100个时期和批量大小等于8,约为40秒,运行在具有单个5950x CPU、128 Gb RAM 和 GTX 3060 GPU 的 Linux 工作站上。生成输出 y 的真实预测器是可访问的。我们使用 PR-AUC 曲线下面积来评估对代谢物和特征的估计重要性的准确性,这在真实预测器的比例较小时是合适的。此外,计算了测试数据类标签预测的准确率,以及特征-代谢物匹配预测的准确率。
选择和预测精度
模拟结果总结如图5所示。在每一行中,前三个图是真实预测器比例为0.10的结果,而最后三个图对应真实预测器比例为0.2的情况。不同的子图呈现了具有各种泊松参数的情况,如每个子图顶部所示。图5a和b显示了在不同设置下代谢物和特征选择的PR-AUC。PR曲线下面积(PR-AUC)通过测量分类模型在识别正例时在平衡精度和召回方面的能力,同时避免假正例,提供了对分类模型性能的全面评估。在我们的研究中,当真实代谢预测器的比例为10%,代谢物数量为1000时,代谢物PR-AUC范围为0.2到0.4,特征PR-AUC范围为0.2到0.35。类似地,对于20%的比例和1000的代谢物数量,代谢物PR-AUC范围为0.4到0.5,特征PR-AUC范围为0.35到0.5。将代谢物数量增加到2000会产生代谢物PR-AUC在0.1到0.2之间,对于10%的比例,特征PR-AUC在0.17到0.3之间,而对于20%的有效比例,代谢物PR-AUC在0.2到0.3之间,特征PR-AUC在0.35到0.45之间。在有效和无效特征之间存在显著不平衡的情况下,对于该度量达到其上限值是具有挑战性的。为了提供一个参考点,处于纯随机条件下,对于固定数量的正特征为100,期望的PR-AUC值分别为1000和2000个特征分别为0.1和0.05。同样,当正特征数量固定为200时,对于1000和2000个特征,平均PR-AUC值分别为0.2和0.1。代谢网络规模的增加,而保持特征数量不变,导致性能下降,这是预期的,因为有更多的噪声代谢物,而分配给每个真实代谢物的真实信号更低。
Figure 5c展示了该方法识别的特征-代谢物匹配的准确性。在不同设置中,识别代谢物和特征之间的真实匹配的准确性在0.61到0.75之间,泊松参数(P_r)有所变化。有趣的是,当P_r较小时,我们观察到代谢网络的两个规模之间存在相反的趋势。当泊松参数较高时,建立了更多特征和代谢物之间的潜在匹配。在较大的代谢网络(2000个代谢物)中,每个代谢物的潜在连接较小,使得特征-代谢物连接选择相对较容易,与存在1000个代谢物的情况相比。总体而言,在多匹配的情况下识别真实匹配以及识别真实贡献的代谢物是一项具有挑战性的任务。仍然有很大的改进空间。然后,我们关注了预测性能。
然后,我们关注模型的预测能力。我们将我们的方法与梯度提升机(GBM)进行了比较,GBM是一种领先的机器学习方法,不考虑特征-代谢物匹配,直接使用特征进行预测。图5d和e显示了在具有1000和2000个代谢物的测试集上的分类准确度。在大多数实验中,使用我们的方法的测试准确度在0.80到0.90之间,表明我们的预测模型具有很好的学习和分类能力。在大多数情况下,我们的方法在测试准确度方面优于GBM,表明其出色的分类能力。总体而言,模拟结果显示我们的方法在解决匹配不确定性问题的同时能够实现良好的分类性能。
特征选择的再现性
关于特征选择的可重复性和稳健性,我们对所选的代谢物在模型训练的不同重复中是否稳定进行了探讨。为了探索这一特性,我们模拟了一个包含4000个样本、1000个特征和1000个代谢物的数据集,其中有100个真实的代谢物通过 logistic 连接函数确定类别 y。实验中使用的泊松率为1,其他参数与先前的模拟实验一致。我们重复了十次训练,并每次记录了前100个代谢物。在这10组前100个最重要的代谢物中,有62个代谢物出现了超过七次,其中40个是真正的预测代谢物。被选中超过五次的代谢物数量为104个,其中64个是真正的预测代谢物。这些结果反映了该方法在不同运行之间具有良好的可重复性。
消融研究
为了验证我们设计的图嵌入在隐藏层中的有效性,我们进行了几个实验,使用一个具有随机打乱边的代谢网络。我们将代谢物大小设置为1000,所有其他参数与先前的模拟研究保持一致。我们通过边交换随机重排代谢网络,从而保持图的度分布不变。如图6d所示,测试准确性保持相似,而当代谢网络被随机重排时,特征和代谢物选择性能显著下降(图6a和b)。如预期所示,当代谢网络被随机重排时,模型无法估计特征和代谢物之间的真实匹配(图6c)。通过与不提供信息的图进行比较,我们展示了我们设计的图嵌入在隐藏层中能够有效捕捉代谢网络的潜在结构,从而提高了特征选择性能。我们的结果表明图嵌入在准确表征代谢网络方面的重要性。我们进一步调查了涉及图嵌入层的不同结构设置的影响。结果总结在补充材料中。
工具和实施
为了方便采用我们的方法,我们开发了一个名为 "meta_matching_tool" 的 Python 软件包,可以从 GitHub 下载并在本地安装。有关 "meta_matching_tool" 的详细教程也可以在我们的 GitHub 仓库 https://github.com/tianlq-prog/SPARSENN 上找到。
该软件包包括两个主要组件:数据预处理和模型训练。模型的输入包括观察到的特征丰度矩阵、每个样本的临床标签、特征到代谢物的匹配矩阵(可选)和代谢网络(可选)。输出包括在测试集上的预测准确性、特征和代谢物的重要性,以及推断出的特征到代谢物的匹配。
对于两个可选的输入,我们提供了与 KEGG 代谢物和 KEGG 代谢网络的匹配。但是,我们注意到原始的非靶向代谢数据需要进行数据预处理。尽管用户可以使用我们 Python 软件包中的函数执行注释过程,并利用内置的 KEGG 网络,但由于不同的数据可能需要不同的预处理过程,我们建议用户根据实际情况调整预处理以更好地适应其数据。
结论
在这项研究中,我们提出了一个用于分析非靶向 LC/MS 代谢数据的统一框架,可以同时解决匹配不确定性问题并进行特征/代谢物选择。该模型在稀疏神经网络的隐藏层中嵌入了特征-代谢物匹配关系和代谢网络。该方法在识别真正有预测性的代谢物和建立正确的代谢物-特征匹配方面表现出很好的性能。我们的模型在两个真实世界的应用中产生了与生物相关的选定代谢物和代谢子网络,突显了我们方法的实际价值。
需要注意的是,我们的框架依赖于已知代谢网络的可用性,其可解释性受限于提供的代谢网络的范围。尽管如此,我们的研究是一次开创性的尝试,因为我们是第一个成功将匹配不确定性问题的解决方案和特征/代谢物选择整合到代谢网络分析和预测建模中的团队。通过这样做,我们的框架为非靶向代谢组学数据的分析提供了统一而全面的方法。除了方法学的进步,我们还开发了一个用户友好的 Python 包,名为 'meta_matching_tool',旨在简化数据预处理和模型训练。
然而,我们承认当前版本的工具存在一些局限性。首先,在数据预处理期间,可用的参考来源目前仅限于KEGG数据库和KEGG代谢网络。虽然它们提供了坚实的基础,但在特定情况下,涵盖更广泛代谢途径的其他数据库可能更适合。
其次,在处理样本量有限的研究时,我们的模型可能需要仔细调整。由于模型依赖于神经网络,样本量和初始随机化可能会影响最终性能。因此,可能需要基于数据的特定特征进行定制的训练技术。
第三,解决匹配不确定性本质上是一个困难的问题。在计算模型中解决它只考虑了潜在可用信息的一部分。此外,这个问题是高维的,即使在仿真中样本量达到数千个,仍然非常有限。匹配不确定性很难仅通过模型化预测中的特征行为来解决,就像我们目前的方法一样。我们的框架尚不支持对部分注释数据集的分析。结合当前的方法与其他特征特性以及部分注释可能会提高性能,并对结果进行更全面的评估。未来的工作将涉及将框架扩展到包括部分注释的数据集,并整合数据相关的采集和DIA数据。
要点
• 我们在该领域开发了一种新颖的方法,能够同时进行预测、代谢物选择,并估计LC/MS特征-代谢物匹配的置信度。
• 这种新方法集成了代谢网络,以选择重要的子网络并实现高度可解释性。
• 这种新方法从两个真实数据集中选择了信息丰富的子网络,展示了它在分析非靶向代谢组学数据方面的价值。
图5. 不同参数的模拟结果,包括代谢物大小、样本大小、有效比例和多重匹配的参数。每一列对应于不同参数的结果,其中'E_r'代表有效代谢物的比例,'P_r'代表决定特征和代谢物之间多重匹配规模的泊松分布参数。(A) 代谢物选择的精度-召回(PR)曲线下面积。 (B) 特征选择的精度-召回(PR)曲线下面积。 (C) 预测特征和代谢物之间真实连接的准确性。 (D) 在代谢物大小为1000的测试数据集上使用我们的方法和GBM的分类准确性。 (E) 在代谢物大小为2000的测试数据集上使用我们的方法和GBM的分类准确性。