一起来学孟德尔随机化(Mendelian Randomization)
孟德尔随机化最近实在是太火了,想不关注都不行,最近也花了点时间研究了一下,和大家分享一下,共同学习。

什么是孟德尔随机化?
在19世纪,孟德尔用豌豆花作为实验材料,通过对豌豆花颜色、形状等特征的观察和统计分析,发现了遗传的基本规律,这就是孟德尔定律。不过,孟德尔定律只适用于单基因的遗传性状,并且无法解释复杂的多基因遗传疾病。此外,孟德尔定律也无法解释环境因素对基因表达的影响,以及基因与环境的相互作用。为了解决这个问题,著名统计学家Fisher提出了孟德尔随机化的概念。
孟德尔随机化(Mendelian randomization,简称MR)是一种基于遗传变异的因果推断方法,其基本原理是利用自然界中的随机分配的基因型对表型的影响来推断生物学因素对疾病的影响。

上面都是概念,下面咱们来举个例子说明一下,假设咱们想了解体重(BMI)对冠心病发病的影响。但是对冠心病影响的因素太多啦,比如高血压、高血糖等,我们怎么才能够排除混杂,确定为体重(BMI)对冠心病有影响呢?我们先要先要选定一个工具基因变量M(假设它为M),这个M要和我们研究的变量X相关,和我们结局变量和混杂因素无关。最后我们通过MR分析得出M基因对Y有影响,因为M基因对Y没有直接关联,所以M基因是通过影响暴露因素X而后从而对Y产生影响。这一联成线的过程可以看做是一个中介效应,暴露因素X(体重)就是中介变量,基因M通过对中介变量X的影响达到对Y的影响。(见下图)
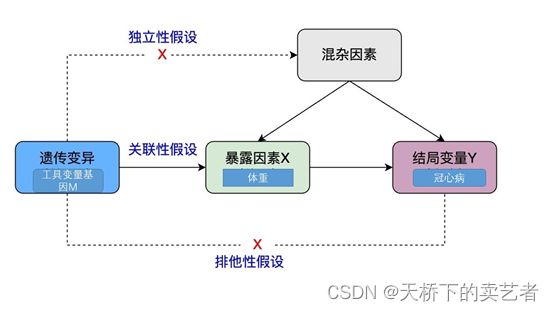
要做孟德尔随机化要有3个假设前提:
- 基因M要和体重(暴露因素)强相关联。(关联性假设)
- 基因M和结局变量冠心病和其他混杂因素没有关联。(独立性假设)
- 基因M只能通过影响体重对冠心病造成影响,不能通过其他途径对冠心病造成影响。(排他性假设)
具体怎么做,首先要用工具,目前主流使用的是TwoSampleMR包,我们需要先安装好,使用代码
devtools::install_github("MRCIEU/TwoSampleMR")
安装TwoSampleMR包会连其他的辅助包一起安装好,安装的时候,有时候Rstudio会提示你是不是要进行其他包的更新,选3不更新就行。
或者使用官方的安装代码
install.packages("remotes")
remotes::install_github("MRCIEU/TwoSampleMR")
安装好以后就可以开始:
第一步:就是获得暴露因素X的SNP数据,(这里就是体重BMI的SNP数据),还有结局冠心病的SNP数据。通常通过各种GWAS数据库或者GWAS文献找到。我们这里使用(https://gwas.mrcieu.ac.uk/datasets/)这个数据库的,它的好处就是直接可以通过TwoSampleMR包下载。下面是它的界面

我们在Trait contains:这里填入体重指数,然后筛选出很多的数据,这里我选择了ieu-a-835这个数据
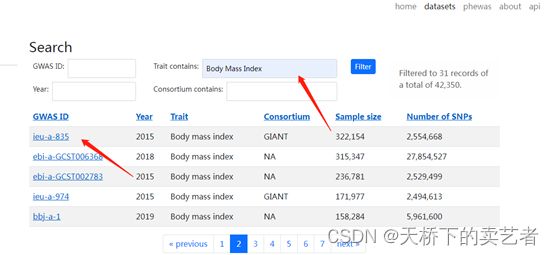
然后我们再找冠心病相关的数据,这里选的是ieu-a-7这个数据

所以,我们需要下载ieu-a-835和ieu-a-7这两个数据。
我们通过TwoSampleMR包把它下载,先使用extract_instruments函数对暴露数据(就是体重BMI的SNP数据)ieu-a-835进行下载,如果是已经下载到电脑里面的数据,我们使用read_exposure_data函数和clump_data函数读取。
library(TwoSampleMR)
exposure_dat <-extract_instruments(outcomes='ieu-a-835',clump=TRUE, r2=0.001,kb=10000,access_token= NULL)
其实clump=TRUE, r2=0.001,kb=10000,access_token= NULL这些都是默认的,你写成这样也是可以的
exposure_dat <-extract_instruments(outcomes='ieu-a-835')

但是这些参数还是很有必要了解一下,p1设置为很小是为了找到与暴露(这里就是与体重BMI相关的SNP),clump=TRUE, r2=0.001,kb=10000这3个指标是为了去掉连锁不平衡的。
运行成功后就生成了exposure_dat文件
第二部:生成exposure_dat文件后第一步就算完成了,接下来我们先看看exposure_dat的SNP数据,共有65个SNP
exposure_dat$SNP

因为ieu-a-7这个数据是冠心病数据,所以这一步要在ieu-a-7这个数据中找到与上面65个SNP匹配的数据,这样就生成了暴露数据,
outcome_dat<-extract_outcome_data(snps=exposure_dat$SNP,
outcomes='ieu-a-7',
proxies = FALSE,
maf_threshold = 0.01,
access_token = NULL)
后面三个参数也是默认的,你写成这样也是可以的
outcome_dat <-extract_outcome_data(snps=exposure_dat$SNP, outcomes="ieu-a-7")
我们来看看参数的解释

第三部:效应等位与效应量保持统一,这一步是必须的,
dat <- harmonise_data(
exposure_dat = exposure_dat,
outcome_dat = outcome_dat
)
这三步走完就可以进行MR分析了,代码非常简单。mr默认使用五种方法( MR Egger,Weighted median,Inverse variance weighted,Simple mode ,Weighted mode )
res <- mr(dat)
res

b是效应值,se是标准误,pval是P值,最重要的就是看Inverse variance weighted这个方法的P值。这里P值小于0.05,表明体重和冠心病病结局是与统计意义的。
接下来进行敏感性分析
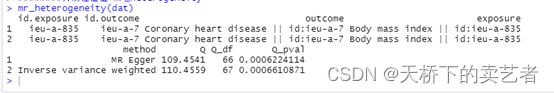
异质性检验 mr_heterogeneity,我这里是存在异质性的
mr_heterogeneity(dat)
水平多效性检验,如果变量工具不通过暴露影响结果,就违反了孟德尔的假设,就是存在多水平效应。
mr_pleiotropy_test(dat)

Leave-one-out analysis,Leave-one-out analysis是指逐步剔除SNP后观察剩余的稳定性,理想的是剔除后变化不大,这和我们的meta分析剔除法很相似。
res_loo <- mr_leaveoneout(dat)
mr_leaveoneout_plot(res_loo)

可视化:
散点图,可以看出趋势是正相关。
p1 <- mr_scatter_plot(res, dat)
p1

森林图,森林图主要是看总效应
res_single <- mr_singlesnp(dat)
mr_forest_plot(res_single)
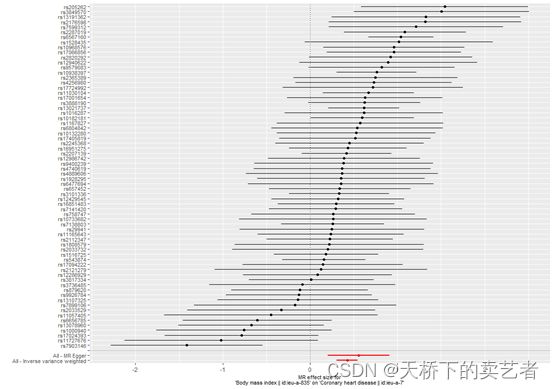
我们可以通过res_single的数据进行提取

然后通过常规方法绘制下面这样的森林图,我已经有很多文章介绍了
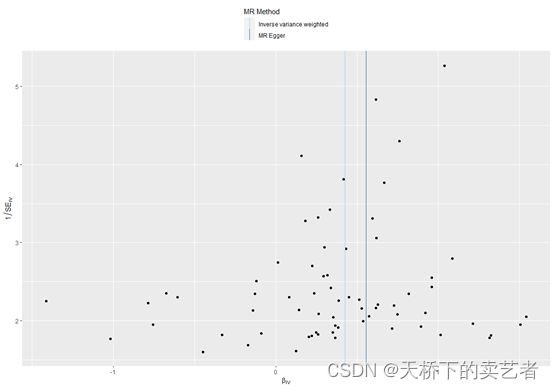
漏斗图
mr_funnel_plot(res_single)
参考文献:
- TwoSampleMR包说明
- https://zhuanlan.zhihu.com/p/433138187
- https://zhuanlan.zhihu.com/p/564866251?utm_id=0
- https://mp.weixin.qq.com/s/PVPO1s7xWwQ9KH1Yk07CaQ
- https://www.frontiersin.org/articles/10.3389/fendo.2023.1125427/full#h11
- https://mp.weixin.qq.com/s/SzctCiipij3_7uICKY61Ug
- https://www.jianshu.com/p/58c7d8541c84