空间转录组分析流程(使用Seurat对空间数据集进行分析)
空间转录组分析流程(使用Seurat对空间数据集进行分析)
因为每次打开这个网页都非常慢,所以我讲这个网页进行一个翻译,方便学习。
使用Seurat对空间数据集进行分析,可视化和集成
1、介绍
本教程演示了如何使用Seurat(> = 3.2)分析空间分布的RNA-seq数据。尽管分析流程类似于单细胞RNA序列分析的Seurat工作流程,但我们引入了更新的交互作用和可视化工具,特别着重于空间和分子信息的整合。本教程将涵盖以下任务,我们认为这些任务在许多空间分析中都是常见的:
- Normalization标准化
- Dimensional reduction and clustering降维和聚类
- Detecting spatially-variable features检测空间可变特征
- Interactive visualization交互可视化
- Integration with single-cell RNA-seq data与单细胞RNA-seq数据整合
- Working with multiple slices处理多个切片
对于我们的第一步,我们分析了使用10x Genomics的Visium技术生成的数据集。我们将扩展Seurat以便在不久的将来使用其他数据类型,包括SLIDE-Seq,STARmap和MERFISH。
首先,我们加载Seurat和此步骤所需的其他软件包。
library("Seurat")
library("SeuratData")
library("ggplot2")
library("patchwork")
library("dplyr")
2、10x Visium
2.1 数据集
在这里,我们将使用使用Visium v1化学方法生成的最近发布的矢状小鼠大脑切片的数据集。有两个连续的前节和两个(匹配的)连续后节。
您可以在此处下载数据,然后使用该Load10X_Spatial()功能将其加载到Seurat中。这将读取spaceranger管道的输出,并返回一个Seurat对象,该对象包含点级表达数据以及组织切片的关联图像。您还可以使用我们的SeuratData包来轻松访问数据,如下所示。安装数据集后,您可以键入*?stxBrain*以了解更多信息。
InstallData("stxBrain")
brain <- LoadData("stxBrain", type = "anterior1")
如何在Seurat中存储空间数据?
来自10x的visium数据包含以下数据类型:
- 按基因表达矩阵的点
- 组织切片的图像(从数据采集期间的H&E染色获得)
- 将原始高分辨率图像与此处用于可视化的较低分辨率图像相关联的缩放因子。
在Seurat对象中,按基因表达矩阵的斑点类似于典型的“ RNA”Assay,但包含斑点水平,而不是单细胞水平数据。图像本身存储在Seurat对象的新images槽中。所述images槽还存储必要的与组织图像上的物理位置相关联的斑点的信息。
2.2 数据预处理
我们通过基因表达数据当场执行的初始预处理步骤与典型的scRNA-seq实验相似。首先,我们需要对数据进行归一化,以解决各个数据点之间的测序深度差异。我们注意到,对于空间数据集,分子计数/斑点的变化可能很大,尤其是在整个组织中细胞密度存在差异的情况下。我们在这里看到大量的异质性,这需要有效的规范化。
plot1 <- VlnPlot(brain, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()
plot2 <- SpatialFeaturePlot(brain, features = "nCount_Spatial") + theme(legend.position = "right")
wrap_plots(plot1, plot2)
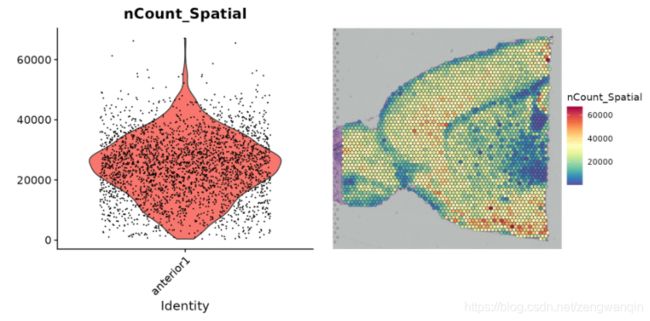
这些图表明,斑点上分子计数的变化不仅是技术上的问题,而且还取决于组织的解剖结构。例如,神经元(例如皮质白质)耗竭的组织区域可再现地显示出较低的分子数。结果,在标准化后强制每个数据点具有相同的底层“大小”的标准方法(例如LogNormalize()函数)可能会出现问题。
作为替代方案,我们建议使用sctransform(Hafemeister和Satija,Genome Biology 2019),它构建了基因表达的正则化负二项式模型,以便在保留生物学差异的同时考虑技术伪像。有关sctransform更多详细信息,请参见文章和Seurat vignette。sctransform可以对数据进行归一化,检测高变异特征并将数据存储在SCT中。
brain <- SCTransform(brain, assay = "Spatial", verbose = FALSE)
2.3基因表达可视化
在Seurat中,我们具有探索空间数据固有的视觉本质并与之交互的功能。SpatialFeaturePlot()在Seurat中的功能扩展为FeaturePlot(),并且可以在组织组织学之上叠加分子数据。例如,在此小鼠大脑数据集中,基因Hpca是强海马标志物,而Ttr是脉络丛的标志物。
SpatialFeaturePlot(brain, features = c("Hpca", "Ttr"))

Seurat中的默认参数强调分子数据的可视化。但是,您还可以通过更改以下参数来调整斑点的大小(及其透明度),以改善组织学图像的可视化:
- pt.size.factor这将缩放斑点的大小。默认值为1.6
- alpha-最小和最大透明度。默认值为c(1,1)。
- 尝试将其设置为alphac(0.1,1),以降低具有较低表达式的点的透明度
p1 <- SpatialFeaturePlot(brain, features = "Ttr", pt.size.factor = 1)
p2 <- SpatialFeaturePlot(brain, features = "Ttr", alpha = c(0.1, 1))
p1 + p2

2.4 降维,聚类和可视化
然后,我们可以使用与scRNA-seq分析相同的工作流程,对RNA表达数据进行降维和聚类。
brain <- RunPCA(brain, assay = "SCT", verbose = FALSE)
brain <- FindNeighbors(brain, reduction = "pca", dims = 1:30)
brain <- FindClusters(brain, verbose = FALSE)
brain <- RunUMAP(brain, reduction = "pca", dims = 1:30)
然后,我们可以在UMAP空间(使用DimPlot())中可视化聚类的结果,或者使用覆盖可视化图像上的聚类结果SpatialDimPlot()。
p1 <- DimPlot(brain, reduction = "umap", label = TRUE)
p2 <- SpatialDimPlot(brain, label = TRUE, label.size = 3)
p1 + p2
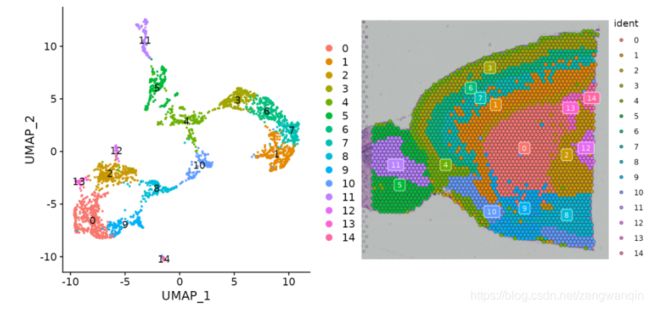
由于存在许多种颜色,因此可视化哪个立体元素属于哪个群集可能具有挑战性。我们有一些策略可以帮助您解决这个问题。设置label参数会在每个群集的中位数处放置一个彩色框(请参见上图)。
您还可以使用cells.highlight参数在上划分特定的关注单元格SpatialDimPlot()。如下所示,这对于区分单个群集的空间定位非常有用。
SpatialDimPlot(brain, cells.highlight = CellsByIdentities(object = brain, idents = c(2, 1, 4, 3,
5, 8)), facet.highlight = TRUE, ncol = 3)

2.5 交互式绘图
我们还内置了许多交互式绘图功能。无论SpatialDimPlot()和SpatialFeaturePlot()都有一个interactive参数,当设置为TRUE,将打开Rstudio观众面板与互动闪亮的情节。下面的示例演示了一种交互式SpatialDimPlot()的方法,您可以在其上悬停并查看单元名称和当前标识类(类似于先前的do.hover行为)。
SpatialDimPlot(brain, interactive = TRUE)
对于SpatialFeaturePlot(),将“交互性”设置为TRUE会弹出一个交互窗格,您可以在其中调整点的透明度,点的大小以及Assay要绘制的和特征。浏览数据后,选择完成按钮将返回最后一个活动图作为ggplot对象。
SpatialFeaturePlot(brain, features = "Ttr", interactive = TRUE)
该LinkedDimPlot()功能将UMAP表示链接到组织图像表示,并允许交互式选择。例如,您可以在UMAP图中选择一个区域,并且图像表示中的相应斑点将突出显示。
LinkedDimPlot(brain)
2.6 识别空间可变特征
Seurat提供了两种工作流程来识别与组织内空间位置相关的分子特征。第一种是基于组织内预先标注的解剖区域执行差异表达,这可以从无监督的聚类或先验知识中确定。在这种情况下,此策略将起作用,因为上面的群集显示出明显的空间限制。
de_markers <- FindMarkers(brain, ident.1 = 5, ident.2 = 6)
SpatialFeaturePlot(object = brain, features = rownames(de_markers)[1:3], alpha = c(0.1, 1), ncol = 3)
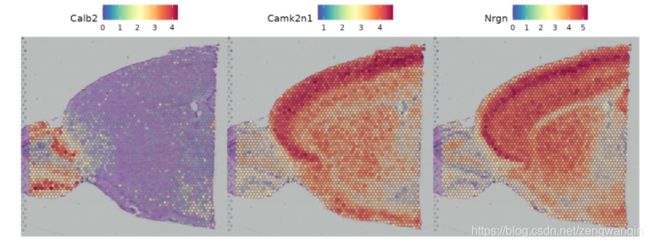
在实施中的另一种方法FindSpatiallyVariables()是在没有预先注释的情况下搜索表现出空间图案的特征。默认方法(method = 'markvariogram)受Trendsceek启发,该工具将空间转录组学数据建模为标记点过程,并计算“变异函数”,该变异函数可识别表达水平取决于其空间位置的基因。更具体地说,此过程计算gamma(r)值,该值测量相距某个“ r”距离的两个点之间的依赖性。默认情况下,我们在这些分析中使用r值“ 5”,并且仅计算可变基因的这些值(其中变异独立于空间位置而计算)以节省时间。
我们注意到,文献中有多种方法可以完成此任务,包括SpatialDE和Splotch。我们鼓励感兴趣的用户探索这些方法,并希望在不久的将来为其提供支持。
brain <- FindSpatiallyVariableFeatures(brain, assay = "SCT", features = VariableFeatures(brain)[1:1000],
selection.method = "markvariogram")
现在,我们可视化此度量确定的前6个特征的表达。
top.features <- head(SpatiallyVariableFeatures(brain, selection.method = "markvariogram"), 6)
SpatialFeaturePlot(brain, features = top.features, ncol = 3, alpha = c(0.1, 1))

2.7 细分解剖区域
与单细胞对象一样,您可以对对象进行子集化以集中处理数据的子集。在这里,我们大约将额叶皮层子集化。该过程还有助于在下一节中将这些数据与皮质scRNA-seq数据集进行整合。首先,我们获取群集的子集,然后根据精确位置进一步细分。子集化后,我们可以在完整图像或裁剪后的图像上可视化皮质细胞。
cortex <- subset(brain, idents = c(1, 2, 3, 4, 6, 7))
# now remove additional cells, use SpatialDimPlots to visualize what to remove
# SpatialDimPlot(cortex,cells.highlight = WhichCells(cortex, expression = image_imagerow > 400 |
# image_imagecol < 150))
cortex <- subset(cortex, anterior1_imagerow > 400 | anterior1_imagecol < 150, invert = TRUE)
cortex <- subset(cortex, anterior1_imagerow > 275 & anterior1_imagecol > 370, invert = TRUE)
cortex <- subset(cortex, anterior1_imagerow > 250 & anterior1_imagecol > 440, invert = TRUE)
p1 <- SpatialDimPlot(cortex, crop = TRUE, label = TRUE)
p2 <- SpatialDimPlot(cortex, crop = FALSE, label = TRUE, pt.size.factor = 1, label.size = 3)
p1 + p2

2.8 与单细胞数据集成
在〜50um时,来自于病毒测定的斑点将涵盖多个细胞的表达谱。对于可获得scRNA-seq数据的系统列表不断增加,用户可能有兴趣对每个空间体素进行“反卷积”以预测细胞类型的潜在组成。在准备此小插图时,我们使用了参考scRNA-seq数据集测试了多种decovonlution和整合方法使用SMART-Seq2协议生成的来自Allen Institute的约14,000个成年小鼠皮质细胞分类学。我们一直发现使用积分方法(而不是反卷积方法)具有更好的性能,这可能是由于表征空间和单细胞数据集的噪声模型存在很大差异,并且积分方法专门设计为对这些差异具有鲁棒性。因此,我们应用了Seurat v3中引入的基于“锚”的集成工作流,该工作流使注释能够从引用到查询集的概率传输。因此,我们利用sctransform归一化方法遵循此处介绍的标签传输工作流程,但期望开发出新方法来完成此任务。
我们首先加载数据(可在此处下载),对scRNA-seq参考进行预处理,然后执行标签转移。该过程为每个点输出每个scRNA-seq派生类的概率分类。我们将这些预测添加为Seurat对象中的一项新分析。
allen_reference <- readRDS("../data/allen_cortex.rds")
# note that setting ncells=3000 normalizes the full dataset but learns noise models on 3k cells
# this speeds up SCTransform dramatically with no loss in performance
library(dplyr)
allen_reference <- SCTransform(allen_reference, ncells = 3000, verbose = FALSE) %>% RunPCA(verbose = FALSE) %>%
RunUMAP(dims = 1:30)
# After subsetting, we renormalize cortex
cortex <- SCTransform(cortex, assay = "Spatial", verbose = FALSE) %>% RunPCA(verbose = FALSE)
# the annotation is stored in the 'subclass' column of object metadata
DimPlot(allen_reference, group.by = "subclass", label = TRUE)

anchors <- FindTransferAnchors(reference = allen_reference, query = cortex, normalization.method = "SCT")
predictions.assay <- TransferData(anchorset = anchors, refdata = allen_reference$subclass, prediction.assay = TRUE,
weight.reduction = cortex[["pca"]], dims = 1:30)
cortex[["predictions"]] <- predictions.assay
现在,我们获得了每个班级每个地点的预测分数。在额叶皮层区域中特别令人感兴趣的是层状兴奋性神经元。在这里,我们可以区分这些神经元亚型的不同顺序层,例如:
DefaultAssay(cortex) <- "predictions"
SpatialFeaturePlot(cortex, features = c("L2/3 IT", "L4"), pt.size.factor = 1.6, ncol = 2, crop = TRUE)

基于这些预测分数,我们还可以预测位置受空间限制的单元格类型。我们使用基于标记点过程的相同方法来定义空间可变特征,但将细胞类型预测得分用作“标记”而不是基因表达。
c
ortex <- FindSpatiallyVariableFeatures(cortex, assay = "predictions", selection.method = "markvariogram",
features = rownames(cortex), r.metric = 5, slot = "data")
top.clusters <- head(SpatiallyVariableFeatures(cortex), 4)
SpatialPlot(object = cortex, features = top.clusters, ncol = 2)

最后,我们证明我们的整合程序能够恢复神经元和非神经元子集(包括层状兴奋性,第1层星形胶质细胞和皮质灰质)的已知空间定位模式。
SpatialFeaturePlot(cortex, features = c("Astro", "L2/3 IT", "L4", "L5 PT", "L5 IT", "L6 CT", "L6 IT",
"L6b", "Oligo"), pt.size.factor = 1, ncol = 2, crop = FALSE, alpha = c(0.1, 1))


2.9 在Seurat中处理多个切片
小鼠大脑的该数据集包含与大脑另一半相对应的另一个切片。在这里,我们将其读入并执行相同的初始归一化。
brain2 <- LoadData("stxBrain", type = "posterior1")
brain2 <- SCTransform(brain2, assay = "Spatial", verbose = FALSE)
为了在同一个Seurat对象中使用多个切片,我们提供了该merge功能。
brain.merge <- merge(brain, brain2)
然后,这使得联合维数减少并在基础RNA表达数据上聚类。
DefaultAssay(brain.merge) <- "SCT"
VariableFeatures(brain.merge) <- c(VariableFeatures(brain), VariableFeatures(brain2))
brain.merge <- RunPCA(brain.merge, verbose = FALSE)
brain.merge <- FindNeighbors(brain.merge, dims = 1:30)
brain.merge <- FindClusters(brain.merge, verbose = FALSE)
brain.merge <- RunUMAP(brain.merge, dims = 1:30)
最后,可以在单个UMAP图中共同可视化数据。SpatialDimPlot()并SpatialFeaturePlot()默认将所有切片绘制为列,将分组/功能绘制为行。
DimPlot(brain.merge, reduction = "umap", group.by = c("ident", "orig.ident"))

SpatialDimPlot(brain.merge)

SpatialFeaturePlot(brain.merge, features = c("Hpca", "Plp1"))
