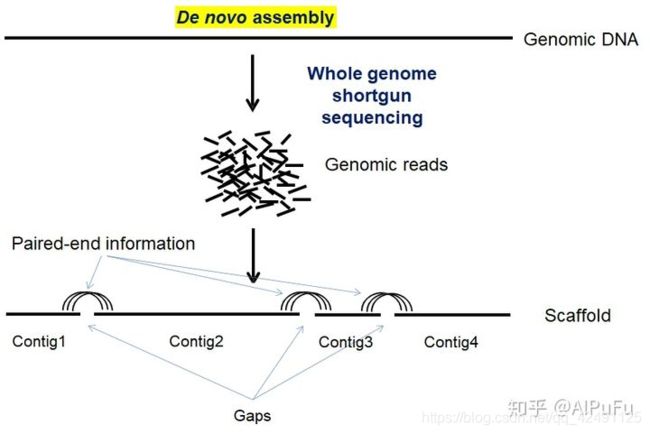
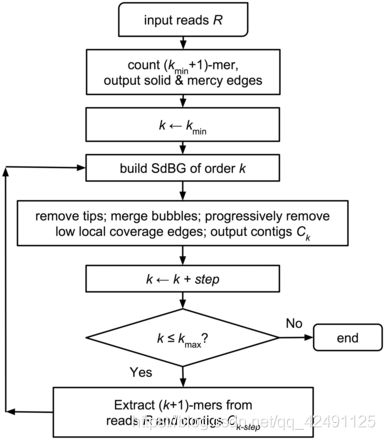
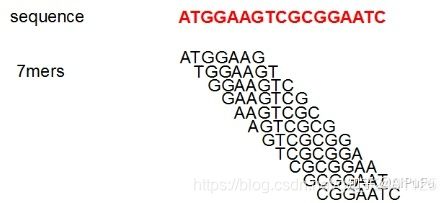
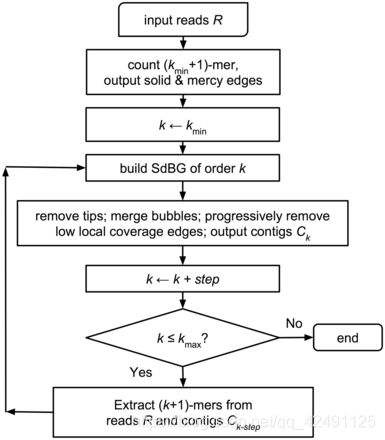
- 上位机知识篇---SD卡&U盘镜像
常用的镜像烧录软件balenaEtcherbalenaEtcher是一个开源的、跨平台的工具,用于将操作系统镜像文件(如ISO和IMG文件)烧录到SD卡和USB驱动器中。以下是其使用方法、使用场景和使用注意事项的介绍:使用方法下载安装:根据自己的操作系统,从官方网站下载对应的安装包。Windows系统下载.exe文件后双击安装;Linux系统若下载的是.deb文件,可在终端执行“sudodpkg-
- php SPOF
贵哥的编程之路(热爱分享 为后来者)
PHP语言经典程序100题php开发语言
1.什么是单点故障(SPOF)?单点故障指的是系统中某个组件一旦失效,整个系统或服务就会不可用。常见的单点有:数据库、缓存、Web服务器、负载均衡、网络设备等。2.常见单点故障场景只有一台数据库服务器,宕机后所有业务不可用只有一台Redis缓存,挂掉后缓存全部失效只有一台Web服务器,挂掉后网站无法访问只有一个负载均衡节点,挂掉后流量无法分发只有一条网络链路,断开后所有服务失联3.消除单点故障的主
- php 高并发下日志量巨大,如何高效采集、存储、分析
贵哥的编程之路(热爱分享 为后来者)
PHP语言经典程序100题php开发语言
1.问题背景高并发系统每秒产生大量日志(如访问日志、错误日志、业务日志等)。单机写入、存储、分析能力有限,容易成为瓶颈。需要支持实时采集、分布式存储、快速检索与分析。2.主流架构方案一、分布式日志采集架构[应用服务器(PHP等)]|v[日志采集Agent(如Filebeat、Fluentd、Logstash)]|v[消息队列/缓冲(如Kafka、Redis、RabbitMQ)]|v[日志存储(如E
- Vue3+Vite+TS+Axios整合详细教程
老马聊技术
VueViteTSvue.js
1.Vite简介Vite是新一代的前端构建工具,在尤雨溪开发Vue3.0的时候诞生。类似于Webpack+Webpack-dev-server。其主要利用浏览器ESM特性导入组织代码,在服务器端按需编译返回,完全跳过了打包这个概念,服务器随起随用。生产中利用Rollup作为打包工具,号称下一代的前端构建工具。vite是一种新型的前端构建工具,能够显著的提升前端开发者的体验。它主要有俩部分组成:一个
- Linux/Centos7离线安装并配置MySQL 5.7
有事开摆无事百杜同学
LInux/CentOS7linuxmysql运维
Linux/Centos7离线安装并配置MySQL5.7超详细教程一、环境准备1.下载MySQL5.7离线包2.使用rpm工具卸载MariaDB(避免冲突)3.创建系统级别的MySQL专用用户二、安装与配置1.解压并重命名MySQL目录2.创建数据目录和配置文件3.设置目录权限4.初始化MySQL5.配置启动脚本6.配置环境变量三、启动与验证1.启动MySQL服务2.获取初始密码3.登录并修改密码
- Linux操作系统磁盘管理
CZZDg
linux运维服务器
目录一.硬盘介绍1.硬盘的物理结构2.CHS编号3.磁盘存储划分4.开机流程5.要点6.磁盘存储数据的形式二.Linux文件系统1.根文件系统2.虚拟文件系统3.真文件系统4.伪文件系统三.磁盘分区与挂载1.磁盘分区方式2.分区命令3.查看与识别命令4.格式化命令5.挂载命令四.LVM逻辑卷1.概述2.管理命令五.磁盘配额1.概述usrquota:支持对用户的磁盘配额grpquota:支持对组的磁
- tcpdump交叉编译
weixin_45673259
tcpdump测试工具网络
1.下载路径官网:https://www.tcpdump.org/2.编译解压:tar-xflibpcap-1.10.4.tar.xztar-xftcpdump-4.99.4.tar.xz编译libpcap./configure--host=mips-v720s229-linux--target=mips-v720s229-linuxCC=/opt/A1/mips-gcc720-uclibc229
- 小林渗透入门:burpsuite+proxifier抓取小程序流量
ξ流ぁ星ぷ132
小程序web安全安全性测试网络安全安全
目录前提:代理:proxifier:步骤:bp证书安装bp设置代理端口:proxifier设置规则:proxifier应用规则:结果:前提:在介绍这两个工具具体实现方法之前,有个很重要的技术必须要大概了解才行---代理。代理:个人觉得代理,简而言之,就是在你和服务器中间的一个中间人,来转达信息。那为什么要代理呢,因为这里的burpsuite要抓包,burpsuite只有做为中间代理人才可以进行拦截
- [特殊字符] 实时数据洪流突围战:Flink+Paimon实现毫秒级分析的架构革命(附压测报告)——日均百亿级数据处理成本降低60%的工业级方案
Lucas55555555
flink大数据
引言:流批一体的时代拐点据阿里云2025白皮书显示,实时数据处理需求年增速达240%,但传统Lambda架构资源消耗占比超运维成本的70%。某电商平台借助Flink+Paimon重构实时数仓后,端到端延迟从分钟级压缩至800ms,计算资源节省5.6万核/月。技术红利窗口期:2025年ApachePaimon1.0正式发布,支持秒级快照与湖仓一体,成为替代Iceberg的新范式一、痛点深挖:实时数仓
- 【Linux内核模块】Linux内核模块程序结构
byte轻骑兵
#嵌入式Linux驱动开发实战linux运维服务器
如果你已经写过第一个"HelloWorld"内核模块,可能会好奇:为什么那个几行代码的程序能被内核识别?那些module_init、MODULE_LICENSE到底是什么意思?今天咱们就来扒一扒内核模块的程序结构,搞清楚一个合格的内核模块到底由哪些部分组成,每个部分又承担着什么角色。目录一、内核模块的"骨架":最简化结构解析二、头文件:内核模块的"说明书"2.1最常用的三个头文件2.2按需添加的其
- LVM逻辑卷扩容
目录1.逻辑卷的简介2.逻辑卷的概念3.相关命令4.建立逻辑卷1.逻辑卷的简介1.LVM是逻辑卷管理(LogicalVolumeManager)的简称,它是Linux环境下对磁盘分区进行管理的一种机制,LVM是建立在硬盘和分区之上的一个逻辑层,来提高磁盘分区管理的灵活性。2.LVM最大的特点就是可以对磁盘进行动态管理。使用了LVM管理分区,动态的调整分区的大小,标准分区是做不到的。2.逻辑卷的概念
- Rocky Linux 8.5/CentOS 8 安装Wine
chen_teacher
linux运维服务器
RockyLinux8.5/CentOS8安装Wine首先配置EPEL镜像配置方法安装Wine首先配置EPEL镜像EPEL(ExtraPackagesforEnterpriseLinux),是由FedoraSpecialInterestGroup维护的EnterpriseLinux(RHEL、CentOS)中经常用到的包。下载地址:https://mirrors.aliyun.com/epel/相
- 系统迁移从CentOS7.9到Rocky8.9
我有两台阿里云上的服务器是CentOS7.9,由于CentOS7已经停止支持,后续使用的话会有安全漏洞,所以需要尽快迁移,个人使用的话目前兼容性好的还是RockyLinux8,很多脚本改改就能用了。一、盘点系统和迁移应用查看当前系统发行版版本cat/etc/os-release盘点迁移清单服务器应用部署方式docker镜像来源v1wordpressdockerdockerhubv1zdirdock
- 【Linux内核模块】Linux内核模块简介
byte轻骑兵
#嵌入式Linux驱动开发实战linuxarm开发运维
你是否好奇过,为什么Linux系统可以在不重启的情况下支持新硬件?为什么修改一个驱动程序不需要重新编译整个内核?这一切都离不开Linux的"模块化魔法"——内核模块(KernelModule)。作为Linux内核最灵活的特性之一,内核模块让开发者可以动态扩展内核功能,今天就来揭开这个神秘组件的面纱。目录一、什么是内核模块?1.1先打个比方:给内核装"插件"1.2技术定义:动态加载的内核代码段1.3
- Linux中LVM逻辑卷扩容
在Linux系统中对根目录所在的LVM逻辑卷进行扩容,需要依次完成物理卷扩容➔卷组扩容➔逻辑卷扩容➔文件系统扩容四个步骤。以下是详细操作流程:一、确认当前磁盘和LVM状态#1.查看磁盘空间使用情况df-h/#2.查看块设备及LVM层级关系lsblk#3.查看LVM详细信息(物理卷PV、卷组VG、逻辑卷LV)pvdisplayvgdisplaylvdisplay二、扩容物理卷(PV)场景1:已有未分
- 自动化运维工程师面试题解析【真题】
ZabbixAgent默认监听的端口是A.10050。以下是关键分析:选项排除:C.80是HTTP默认端口,与ZabbixAgent无关。D.5432是PostgreSQL数据库的默认端口,不涉及ZabbixAgent。B.10051是ZabbixServer的默认监听端口,用于接收Agent发送的数据,而非Agent自身的监听端口。ZabbixAgent的配置:根据官方文档,ZabbixAgen
- redis集群之Sentinel哨兵高可用
会飞的爱迪生
redisredissentinelbootstrap
Sentinel是官网推荐的高可用(HA)解决方案,可以实现redis的高可用,即主挂了从代替主工作,在一台单独的服务器上运行多个sentinel,去监控其他服务器上的redismaster-slave状态(可以监控多个master-slave),当发现master宕机后sentinel会在slave中选举并启动新的master。至少需要3台redis才能建立起基于哨兵的reids集群。一、通过s
- 在 Windows 上安装 Docker Desktop
不老刘
人工智能windowsdocker容器
还是简单说一下,如何在Windows上安装DockerDesktop,具体步骤如下:系统要求Windows10/1164-bit(专业版、企业版或教育版,版本21H2或更高)启用WSL2(WindowsSubsystemforLinux2)或Hyper-V至少4GB内存BIOS中启用虚拟化(VT-x/AMD-V)安装步骤1.下载DockerDesktop访问Docker官网下载页面。下载Docke
- .NET 一款基于BGInfo的红队内网渗透工具
dot.Net安全矩阵
网络.net安全.netcoreweb安全矩阵
01阅读须知此文所提供的信息只为网络安全人员对自己所负责的网站、服务器等(包括但不限于)进行检测或维护参考,未经授权请勿利用文章中的技术资料对任何计算机系统进行入侵操作。利用此文所提供的信息而造成的直接或间接后果和损失,均由使用者本人负责。本文所提供的工具仅用于学习,禁止用于其他方面02基本介绍在内网渗透过程中,白名单绕过是红队常见的技术需求。Sharp4Bginfo.exe是一款基于微软签名工具
- 【运维实战】解决 K8s 节点无法拉取 pause:3.6 镜像导致 API Server 启动失败的问题
gs80140
各种问题运维kubernetes容器
目录【运维实战】解决K8s节点无法拉取pause:3.6镜像导致APIServer启动失败的问题问题分析✅解决方案:替代拉取方式导入pause镜像Step1.从私有仓库拉取pause镜像Step2.重新打tag为Kubernetes默认命名Step3.导出镜像为tar包Step4.拷贝镜像到目标节点Step5.在目标节点导入镜像到containerd的k8s.io命名空间Step6.验证镜像是否导
- 【Linux】进程间通信-管道通信实验
会的全对٩(ˊᗜˋ*)و
Linuxlinux经验分享
要求:利用有名管道编写简单的聊天程序,聊天双方在线才能说话,一方说话后需另一方应答才能继续说话,即一来一往的聊天模式,如果输入quit则退出聊天程序。代码实现:进程A#include#include#include#include#include#include#defineFIFO_A"/tmp/chat_fifo_a"//进程A写消息,进程B读消息#defineFIFO_B"/tmp/chat
- 【个人笔记】负载均衡
撰卢
笔记负载均衡运维
文章目录nginx反向代理的好处负载均衡负载均很的配置方式均衡负载的方式nginx反向代理的好处提高访问速度进行负载均衡保证后端服务安全负载均衡负载均衡,就是把大量的请求按照我们指定的方式均衡的分配给集群中的每台服务器负载均很的配置方式upstreamwebservers{server192.168.100.128:8080server192.168.100.129:8080}server{lis
- Centos7安装uwsgi详细步骤
快乐骑行^_^
大数据Centos7安装uwsgi
Centos7安装uwsgi详细步骤步骤一:下载源码到centos7服务器步骤二:解压步骤三:编译环境准备步骤四:进入解压目录,并且编译uwsgi步骤五:准备测试安装是否成功的python代码testUwsgi步骤六:启动uWSGI来运行一个HTTP服务器步骤七:服务器ip+端口号访问步骤一:下载源码到centos7服务器uwsgi最新版2.0.20下载地址如下:https://github.co
- Python uWSGI 安装配置
AI老李
pythonpython开发语言
关键要点uWSGI安装和配置适合PythonWSGI应用,资源丰富,适合初学者和中级用户。推荐菜鸟教程和官方文档,涵盖Linux和Windows环境。配置需注意操作系统差异和框架(如Django、Flask)需求。安装步骤uWSGI安装通常通过pip或源码编译完成。以下是基本步骤:Linux:安装依赖(如build-essentialpython-dev),然后用pipinstalluwsgi或编
- 上位机知识篇---Linux中的文件挂载
Atticus-Orion
上位机操作篇linux运维网络文件挂载
文章目录前言1.挂载的基本概念文件系统挂载点设备文件2.挂载的命令挂载文件系统示例卸载文件系统示例3.挂载的常用选项示例4.自动挂载(/etc/fstab文件)示例使用UUID挂载5.挂载网络文件系统(NFS)挂载NFS示例6.挂载ISO文件挂载ISO文件示例7.查看已挂载的文件系统8.挂载的注意事项9.挂载的常见问题挂载失败卸载失败10.总结前言在Linux系统中,文件挂载是指将一个文件系统(如
- Ubuntu 服务器虚拟主机,ubuntu云服务器虚拟机
Gamer42
Ubuntu服务器虚拟主机
ubuntu云服务器虚拟机内容精选换一换通过云服务器或者外部镜像文件创建私有镜像时,如果云服务器或镜像文件所在虚拟机的网络配置是静态IP地址时,您需要修改网卡属性为DHCP,以使私有镜像发放的新云服务器可以动态获取IP地址。本节以WindowsServer2008R2操作系统为例。其他操作系统配置方法略有区别,请参考对应操作系统的相关资料进行操作,文档中不对此进行详细说明后端虚拟机绑定EIP。登录
- 《C++性能优化指南》 linux版代码及原理解读 第一章
v俊逸
C++性能优化指南性能优化C++性能优化性能优化
概述:目录概述:性能优化的必要性:C++代码优化策略总结用好的编译器并用好编译器使用更好的算法使用更好的库减少内存分配和复制移除计算使用更好的数据结构提高并发性优化内存管理性能优化的必要性:按照当今的CPU运行速度来说,执行一条指令所需要的时间是10的-9次方的时间单位,如此快速的执行速度是否就没有性能优化的必要了呢?其实不然,性能优化与CPU的执行速度并无非常大的关系,试想一下,一段代码,如果用
- 《C++性能优化指南》 linux版代码及原理解读 第四章
v俊逸
C++性能优化指南性能优化C++性能优化指南性能优化
目录概述为什么字符串很麻烦字符串是动态分配的字符串赋值背后的操作如何面对字符串会进行大量复制写时复制COW(copyonwrite)尝试优化字符串避免临时字符串通过预留存储空间减少内存分配通过传递引用减少实参复制使用迭代器操作减少循环中的比较操作减少返回值的复制还没有结束,使用字符数组代替字符串再次优化字符串尝试其他的算法叠加以前的优化方式使用其他的编译器使用其他字符串的库功能丰富的字符串库使用s
- 如何在 Linux 上安装 RTX 5090 / 5080 /5070 Ti / 5070 驱动程序 — 详细指南
知识大胖
NVIDIAGPU和大语言模型开发教程linux运维服务器
简介为了获得最佳性能,您需要在Linux上运行5090/5080/5070Ti/5070或其他50系列GPU(或Windows上的WSL)。这篇文章将包含有关如何操作的详细指南。主线内核和驱动程序怪癖之旅Nvidia50系列GPU拥有最新的Nvidia技术。但是,新硬件需要一些新软件或更新,这需要一些耐心。如果您在这里,您可能会遇到Ubuntu默认设置的障碍。不要害怕!我最近自己摸索了这个迷宫,结
- 使用 Ollama 、 DeepSeek和QWEN的模型上下文协议 (MCP) ,使用本地 LLM 教程的 MCP 服务器
知识大胖
NVIDIAGPU和大语言模型开发教程服务器运维人工智能qwen2vldeepseek
简介模型上下文协议:MCP服务器据称是AI领域的下一个重大改变者,它将使AI代理变得比我们想象的更加先进。MCP或模型上下文协议由Anthropic去年发布,它可以帮助LLM连接软件并对其进行控制。但有一个问题大多数MCP服务器都与ClaudeAI兼容,尤其是ClaudeAI桌面应用程序,但它们有自己的限制。有没有办法我们可以使用本地LLM运行MCP服务器?是的,在这个特定的逐步详细教程中,我们将
- [黑洞与暗粒子]没有光的世界
comsci
无论是相对论还是其它现代物理学,都显然有个缺陷,那就是必须有光才能够计算
但是,我相信,在我们的世界和宇宙平面中,肯定存在没有光的世界....
那么,在没有光的世界,光子和其它粒子的规律无法被应用和考察,那么以光速为核心的
&nbs
- jQuery Lazy Load 图片延迟加载
aijuans
jquery
基于 jQuery 的图片延迟加载插件,在用户滚动页面到图片之后才进行加载。
对于有较多的图片的网页,使用图片延迟加载,能有效的提高页面加载速度。
版本:
jQuery v1.4.4+
jQuery Lazy Load v1.7.2
注意事项:
需要真正实现图片延迟加载,必须将真实图片地址写在 data-original 属性中。若 src
- 使用Jodd的优点
Kai_Ge
jodd
1. 简化和统一 controller ,抛弃 extends SimpleFormController ,统一使用 implements Controller 的方式。
2. 简化 JSP 页面的 bind, 不需要一个字段一个字段的绑定。
3. 对 bean 没有任何要求,可以使用任意的 bean 做为 formBean。
使用方法简介
- jpa Query转hibernate Query
120153216
Hibernate
public List<Map> getMapList(String hql,
Map map) {
org.hibernate.Query jpaQuery = entityManager.createQuery(hql);
if (null != map) {
for (String parameter : map.keySet()) {
jp
- Django_Python3添加MySQL/MariaDB支持
2002wmj
mariaDB
现状
首先,
[email protected] 中默认的引擎为 django.db.backends.mysql 。但是在Python3中如果这样写的话,会发现 django.db.backends.mysql 依赖 MySQLdb[5] ,而 MySQLdb 又不兼容 Python3 于是要找一种新的方式来继续使用MySQL。 MySQL官方的方案
首先据MySQL文档[3]说,自从MySQL
- 在SQLSERVER中查找消耗IO最多的SQL
357029540
SQL Server
返回做IO数目最多的50条语句以及它们的执行计划。
select top 50
(total_logical_reads/execution_count) as avg_logical_reads,
(total_logical_writes/execution_count) as avg_logical_writes,
(tot
- spring UnChecked 异常 官方定义!
7454103
spring
如果你接触过spring的 事物管理!那么你必须明白 spring的 非捕获异常! 即 unchecked 异常! 因为 spring 默认这类异常事物自动回滚!!
public static boolean isCheckedException(Throwable ex)
{
return !(ex instanceof RuntimeExcep
- mongoDB 入门指南、示例
adminjun
javamongodb操作
一、准备工作
1、 下载mongoDB
下载地址:http://www.mongodb.org/downloads
选择合适你的版本
相关文档:http://www.mongodb.org/display/DOCS/Tutorial
2、 安装mongoDB
A、 不解压模式:
将下载下来的mongoDB-xxx.zip打开,找到bin目录,运行mongod.exe就可以启动服务,默
- CUDA 5 Release Candidate Now Available
aijuans
CUDA
The CUDA 5 Release Candidate is now available at http://developer.nvidia.com/<wbr></wbr>cuda/cuda-pre-production. Now applicable to a broader set of algorithms, CUDA 5 has advanced fe
- Essential Studio for WinRT网格控件测评
Axiba
JavaScripthtml5
Essential Studio for WinRT界面控件包含了商业平板应用程序开发中所需的所有控件,如市场上运行速度最快的grid 和chart、地图、RDL报表查看器、丰富的文本查看器及图表等等。同时,该控件还包含了一组独特的库,用于从WinRT应用程序中生成Excel、Word以及PDF格式的文件。此文将对其另外一个强大的控件——网格控件进行专门的测评详述。
网格控件功能
1、
- java 获取windows系统安装的证书或证书链
bewithme
windows
有时需要获取windows系统安装的证书或证书链,比如说你要通过证书来创建java的密钥库 。
有关证书链的解释可以查看此处 。
public static void main(String[] args) {
SunMSCAPI providerMSCAPI = new SunMSCAPI();
S
- NoSQL数据库之Redis数据库管理(set类型和zset类型)
bijian1013
redis数据库NoSQL
4.sets类型
Set是集合,它是string类型的无序集合。set是通过hash table实现的,添加、删除和查找的复杂度都是O(1)。对集合我们可以取并集、交集、差集。通过这些操作我们可以实现sns中的好友推荐和blog的tag功能。
sadd:向名称为key的set中添加元
- 异常捕获何时用Exception,何时用Throwable
bingyingao
用Exception的情况
try {
//可能发生空指针、数组溢出等异常
} catch (Exception e) {
- 【Kafka四】Kakfa伪分布式安装
bit1129
kafka
在http://bit1129.iteye.com/blog/2174791一文中,实现了单Kafka服务器的安装,在Kafka中,每个Kafka服务器称为一个broker。本文简单介绍下,在单机环境下Kafka的伪分布式安装和测试验证 1. 安装步骤
Kafka伪分布式安装的思路跟Zookeeper的伪分布式安装思路完全一样,不过比Zookeeper稍微简单些(不
- Project Euler
bookjovi
haskell
Project Euler是个数学问题求解网站,网站设计的很有意思,有很多problem,在未提交正确答案前不能查看problem的overview,也不能查看关于problem的discussion thread,只能看到现在problem已经被多少人解决了,人数越多往往代表问题越容易。
看看problem 1吧:
Add all the natural num
- Java-Collections Framework学习与总结-ArrayDeque
BrokenDreams
Collections
表、栈和队列是三种基本的数据结构,前面总结的ArrayList和LinkedList可以作为任意一种数据结构来使用,当然由于实现方式的不同,操作的效率也会不同。
这篇要看一下java.util.ArrayDeque。从命名上看
- 读《研磨设计模式》-代码笔记-装饰模式-Decorator
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
import java.io.BufferedOutputStream;
import java.io.DataOutputStream;
import java.io.FileOutputStream;
import java.io.Fi
- Maven学习(一)
chenyu19891124
Maven私服
学习一门技术和工具总得花费一段时间,5月底6月初自己学习了一些工具,maven+Hudson+nexus的搭建,对于maven以前只是听说,顺便再自己的电脑上搭建了一个maven环境,但是完全不了解maven这一强大的构建工具,还有ant也是一个构建工具,但ant就没有maven那么的简单方便,其实简单点说maven是一个运用命令行就能完成构建,测试,打包,发布一系列功
- [原创]JWFD工作流引擎设计----节点匹配搜索算法(用于初步解决条件异步汇聚问题) 补充
comsci
算法工作PHP搜索引擎嵌入式
本文主要介绍在JWFD工作流引擎设计中遇到的一个实际问题的解决方案,请参考我的博文"带条件选择的并行汇聚路由问题"中图例A2描述的情况(http://comsci.iteye.com/blog/339756),我现在把我对图例A2的一个解决方案公布出来,请大家多指点
节点匹配搜索算法(用于解决标准对称流程图条件汇聚点运行控制参数的算法)
需要解决的问题:已知分支
- Linux中用shell获取昨天、明天或多天前的日期
daizj
linuxshell上几年昨天获取上几个月
在Linux中可以通过date命令获取昨天、明天、上个月、下个月、上一年和下一年
# 获取昨天
date -d 'yesterday' # 或 date -d 'last day'
# 获取明天
date -d 'tomorrow' # 或 date -d 'next day'
# 获取上个月
date -d 'last month'
#
- 我所理解的云计算
dongwei_6688
云计算
在刚开始接触到一个概念时,人们往往都会去探寻这个概念的含义,以达到对其有一个感性的认知,在Wikipedia上关于“云计算”是这么定义的,它说:
Cloud computing is a phrase used to describe a variety of computing co
- YII CMenu配置
dcj3sjt126com
yii
Adding id and class names to CMenu
We use the id and htmlOptions to accomplish this. Watch.
//in your view
$this->widget('zii.widgets.CMenu', array(
'id'=>'myMenu',
'items'=>$this-&g
- 设计模式之静态代理与动态代理
come_for_dream
设计模式
静态代理与动态代理
代理模式是java开发中用到的相对比较多的设计模式,其中的思想就是主业务和相关业务分离。所谓的代理设计就是指由一个代理主题来操作真实主题,真实主题执行具体的业务操作,而代理主题负责其他相关业务的处理。比如我们在进行删除操作的时候需要检验一下用户是否登陆,我们可以删除看成主业务,而把检验用户是否登陆看成其相关业务
- 【转】理解Javascript 系列
gcc2ge
JavaScript
理解Javascript_13_执行模型详解
摘要: 在《理解Javascript_12_执行模型浅析》一文中,我们初步的了解了执行上下文与作用域的概念,那么这一篇将深入分析执行上下文的构建过程,了解执行上下文、函数对象、作用域三者之间的关系。函数执行环境简单的代码:当调用say方法时,第一步是创建其执行环境,在创建执行环境的过程中,会按照定义的先后顺序完成一系列操作:1.首先会创建一个
- Subsets II
hcx2013
set
Given a collection of integers that might contain duplicates, nums, return all possible subsets.
Note:
Elements in a subset must be in non-descending order.
The solution set must not conta
- Spring4.1新特性——Spring缓存框架增强
jinnianshilongnian
spring4
目录
Spring4.1新特性——综述
Spring4.1新特性——Spring核心部分及其他
Spring4.1新特性——Spring缓存框架增强
Spring4.1新特性——异步调用和事件机制的异常处理
Spring4.1新特性——数据库集成测试脚本初始化
Spring4.1新特性——Spring MVC增强
Spring4.1新特性——页面自动化测试框架Spring MVC T
- shell嵌套expect执行命令
liyonghui160com
一直都想把expect的操作写到bash脚本里,这样就不用我再写两个脚本来执行了,搞了一下午终于有点小成就,给大家看看吧.
系统:centos 5.x
1.先安装expect
yum -y install expect
2.脚本内容:
cat auto_svn.sh
#!/bin/bash
- Linux实用命令整理
pda158
linux
0. 基本命令 linux 基本命令整理
1. 压缩 解压 tar -zcvf a.tar.gz a #把a压缩成a.tar.gz tar -zxvf a.tar.gz #把a.tar.gz解压成a
2. vim小结 2.1 vim替换 :m,ns/word_1/word_2/gc
- 独立开发人员通向成功的29个小贴士
shoothao
独立开发
概述:本文收集了关于独立开发人员通向成功需要注意的一些东西,对于具体的每个贴士的注解有兴趣的朋友可以查看下面标注的原文地址。
明白你从事独立开发的原因和目的。
保持坚持制定计划的好习惯。
万事开头难,第一份订单是关键。
培养多元化业务技能。
提供卓越的服务和品质。
谨小慎微。
营销是必备技能。
学会组织,有条理的工作才是最有效率的。
“独立
- JAVA中堆栈和内存分配原理
uule
java
1、栈、堆
1.寄存器:最快的存储区, 由编译器根据需求进行分配,我们在程序中无法控制.2. 栈:存放基本类型的变量数据和对象的引用,但对象本身不存放在栈中,而是存放在堆(new 出来的对象)或者常量池中(字符串常量对象存放在常量池中。)3. 堆:存放所有new出来的对象。4. 静态域:存放静态成员(static定义的)5. 常量池:存放字符串常量和基本类型常量(public static f