24分+的医药顶刊带你学习表观组学解析超级热点“肿瘤耐药”的机制
对癌症患者采用治疗干预时获得性耐药是转移性癌症复发的主要原因。此前,获得性耐药发展的研究主要集中在识别耐药肿瘤中常见的基因突变。越来越多的证据表明,在永久性获得性耐药出现之前,癌症中存在一种表观遗传调控的可逆耐药状态,这种可逆状态可能会导致不同的抗性机制。这些癌症中出现组蛋白去甲基化酶的持续表达,给转录适应和耐药性提供可能。因此,靶向表观遗传调控因子是逆转肿瘤耐药的有效策略。
在黑素瘤小鼠模型中对于治疗诱导的可逆细胞重编程以及BRAF抑制剂治疗进展的研究还属于初期。然而,促进癌症持久体从休眠状态转变为增殖状态的分子机制仍有待研究。今天我们和大家分享一篇澳大利亚昆士兰大学Helmut Schaider团队在医药顶刊Drug Resistance Updates (IF :24.3)发表题为《H3K4me3 remodeling induced acquired resistance through O-GlcNAc transferase》的文章,其对获得性耐药肿瘤细胞模型进行H3K4me3 ChIP-Seq和代谢组研究,揭示了获得性性耐药的基本机制。
技术路线

研究结果
1、不同耐药阶段细胞准备与鉴定
诱导耐药细胞(IDTC):肿瘤细胞IDTC(Induction of Drug-Tolerant Cells)指通过药物处理使肿瘤细胞对药物产生耐受性的过程;DTP(Drug-Tolerant Population)是指药物耐受性细胞群体;DTPP(Drug-Tolerant Population Persistence)则是指药物耐受性细胞群体的持久性。癌细胞IDTC是指通过药物处理使癌细胞对药物产生耐受性的过程。耐药性一旦产生,肿瘤细胞就会形成DTP,即药物耐受性细胞群体。DTPP则是指DTP在药物撤离后的持久性,即药物耐受性细胞群体在药物撤离后是否能够继续生长和繁殖。
根据黑色素瘤细胞WM164暴露于BRAF抑制剂达拉菲尼时间的细胞周期情况将其划分为:Parent(0d)、 IDTC(15d)、IDTC colony(45d)、Mature colony(75d)、Resistant(90d)(图1a)。IDTC colony数量在治疗开始后50天左右达到高峰,之后逐渐减少和消失且与癌细胞增殖无关,随着时间推移癌细胞增殖增加(图.1b/c)。由于IDTC colony早期状态的可逆性,作者假设 IDTC colony的耐药状态也是可逆的。
为了验证这一假设,作者将经过45和65天BRAF抑制剂处理后分离出来的WM164细胞进行21天的停药期,后续再次进行BRAF抑制剂处理。将结果与暴露于BRAF抑制剂的WM164 parent和WM164 BRAF抑制剂处理90d进行比较。结果经过45天处理的细胞没有表现出任何耐药性的迹象,而经过65天处理的细胞显示出对BRAF抑制剂的部分耐药性。
IDTC密集排列的细胞结构对增殖的重新启动可能起着重要的作用,将暴露于BRAF抑制剂45、55、65和75天后分离的WM164 IDTC停药21d,再次用BRAF抑制剂刺激,同时将将经过BRAF抑制剂处理75天后的WM164正常集落形态细胞分离出来作为对照;结果发现早期(45和55天),密集细胞群落完全恢复了对BRAF抑制剂的敏感性(图.1e),处理65天的集落显示部分耐药,处理75天的集落显示接近完全耐药(图. 1e),说明75天耐药表型逐渐稳定。相比之下,周围正常分布状态的细胞在处理75天停药处理后仍可恢复药物敏感性。
为了探究研究形成IDTC细胞不同集落的细胞来源,将表达GFP-和RFP-的WM164细胞按1:1的比例共培养,并将其暴露于达拉菲尼。结果25%的GFP、25%RFP和50%双阳性(黄色),表明IDTC密集细胞群落以多细胞融合为主(图.1h)。为了证实这些结果,对未经处理的parent细胞和药物处理75天IDTC colony分离的细胞进行了全染色体染色(mFISH)分析,表明单一的IDTC集落细胞来源于多个parent细胞(图.1i)。
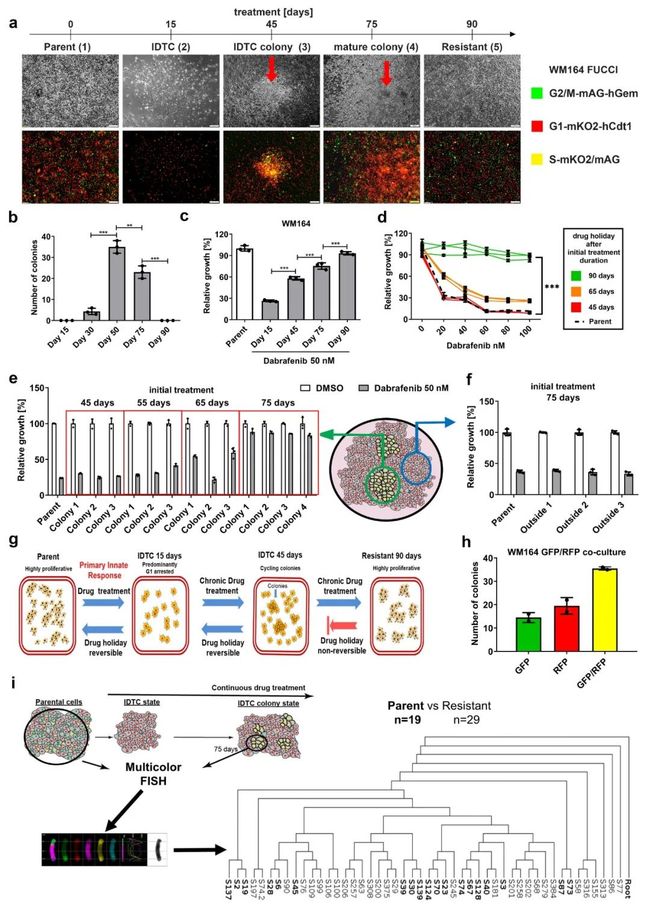
图 1 肿瘤细胞耐药性的获得与发展
2、永久性耐药肿瘤细胞中H3K4me3的重塑变化
之前研究结果显示在多种癌症形成永久耐药性都会存在H3K4me3的下调,因此利用WB检测多种癌细胞系早期IDTC和集落形成后的H3K4me3水平,这些癌细胞系分别用BRAF抑制剂(WM164和A375)、EGFR抑制剂(H1975)和多西他赛(A549)处理。结果表明在处理15天IDTC样品中H3K4me3水平降低,处理15天后H3K4me3水平显著增高。在WM164和A375在处理90天后,耐药细胞H3K4me3水平保持稳定(图.2a)。
为了进一步研究获得性耐药发展过程中H3K4me3重塑对应影响的基因,对未处理的parent细胞、IDTC colony和永久耐药细胞进行染色质免疫共沉淀和高通量测序(ChIP-seq)。ChIP-seq结果显示,未处理parent细胞中有32322个peak,IDTC colony中有22854个peak,耐药细胞中有20087个峰。差异分析显示H3K4me3存在广泛重塑,与parent细胞相比,IDTC colony和耐药细胞中分别有7212和9786个差异peak。选择对应基因进行ChIP-qPCR验证ChIP-seq实验数据。ChIP-seq结果显示H3K4me3标记在转录起始位点(TSS)周围分布(图.2b)。与IDTC colony或耐药细胞相比,parent中分别对应有4766和6548个差异关联基因。这些基因的通路富集分析显示H3K4me3重塑影响最强的通路为:细胞对压力反应、WNT信号相关通路、SUMO化修饰相关通路;而参与细胞周期调控相关通路的基因,p53信号通路和Rho GTPase信号通路显示H3K4me3标记减少。
转录组和ChIP-seq相关性分析结果表明H3K4me3标记与基因表达正相关(Pearson相关性0.42),表观遗传重塑与基因表达相关(图.2d)。差异标记基因的通路富集分析显示,IDTC colony vs parent、耐药vs parent的top富集通路存在大量重叠。尽管IDTC colony和耐药ChIP-seq数据集具有很高的相似性,但直接比较IDTC colony和耐药细胞发现,与IDTC colony相比,耐药细胞中H3K4me3差异标记峰减少了3488个,增加了5503个;通路富集分析表明,与耐药细胞相比,IDTC colony在Rho GTPase信号通路、细胞周期进展、RUNX2信号通路和生物钟相关的通路中H3K4me3增加,而在激酶信号通路和代谢相关的通路中H3K4me3减少(图.2e); 暗示后期获得性药物抗性发展过程中逐渐发生的表观遗传调节适应。
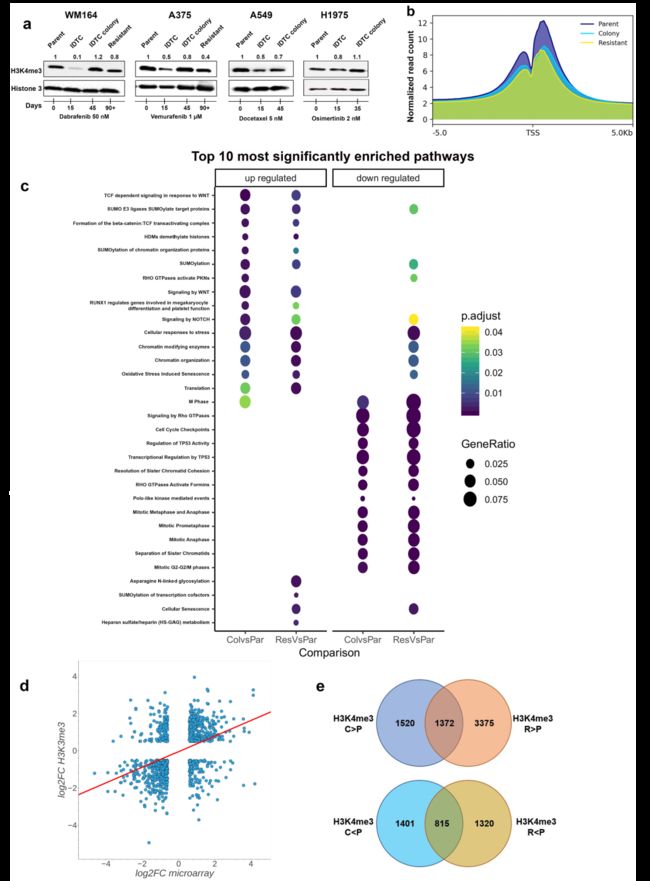
图2 H3K4me3在永久获得性耐药发展过程中的重塑
3、诱导耐药的H3K4me3重塑优先发生在CpG岛上
对H3K4me3 peak进行Motif分析,以确定可能与表观遗传重塑相关的转录因子。在IDTC colony和耐药细胞中motif前20名中最常见的5个是ELK1、ARNT、ELK4、ELF1和FLI1。对H3K4me3差异调控序列进行进一步分析,前20个motif中MYB、BMYB、FLI1、ELK1、ELK4、ETV2和RFX6是7个常见的;表明H3K4me3在ELK1、ELK4和FLI1 DNA结合基序区域显著积累。对parent和IDTC colony H3K4me3差异peak的分析表明,大部分表观遗传变化发生在CpG岛区域。parent和IDTC colony/耐药细胞之间的H3K4me3差异peak与CpG岛重叠率分别为82%(4228/5165)和88%(6361/7249),表明H3K4me3优先在与CpG岛区域重塑(图.3c、3d)。
接下来,作者对先前被认为与获得性耐药相关的21个基因的表达进行检测。在耐药相关基因中,52%(11/21)的H3K4me3 peak的变化在TSS附近,且基因表达(log2FC RT-qPCR)与H3K4me3标记(log2FC ChIP-seq)之间存在很强的相关性(Pearson 's 相关性> 0.95)(图3e)。在所有H3K4me3差异表达的基因中,73%(8/11)的基因表达发生了显著变化(在IDTC菌落状态下,2个基因表达下调,6个基因表达上调)(图.3e)。这表明CpG岛特异性区域H3K4me3的重塑对其调控起着重要作用。作者选择EGFR、CDH2(上调且标记H3K4me3)和PDGFRB(上调且未标记H3K4me3),利用ChIP qPCR验证H3K4me3的富集,进一步确认了ChIP实验的特异性。
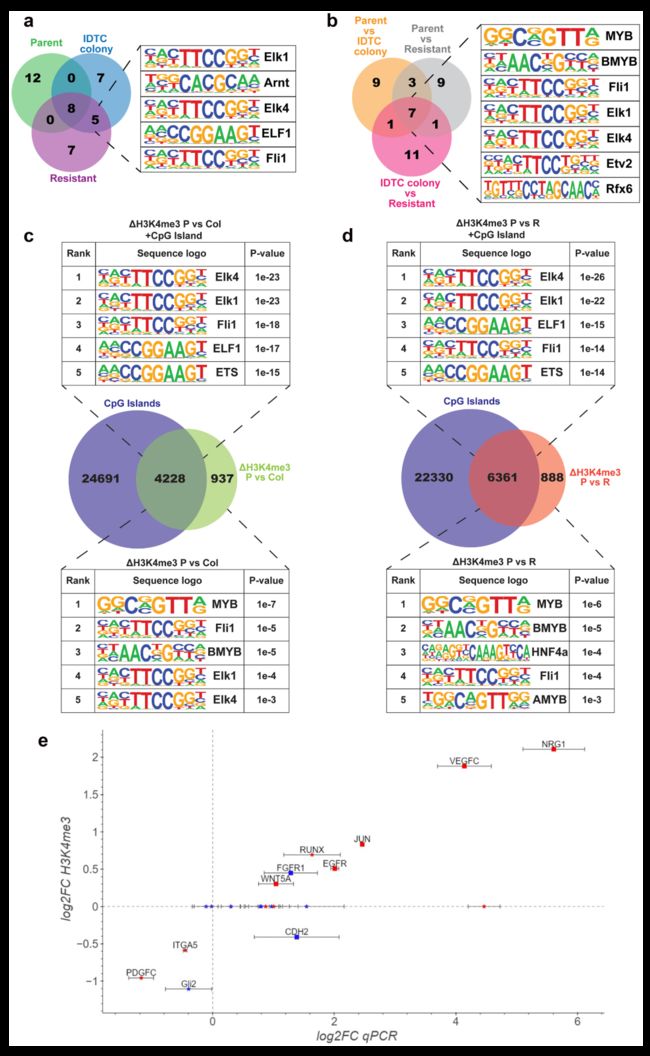
图3 获得性耐药H3K4me3重塑优先发生在CpG岛区域
4、短时耐药IDTC colony通过O-GlcNAc糖基化转移酶上调H3K4me3
由于对CpG岛区域H3K4me3差异标记peak的分析显示,转录因子 ELK1和ELK4是首选motif,没有发现任何相关文献将ELK1和ELK4与H3K4me3上调联系起来。本研究中也没有发现在IDTC colony状态下ELK1或ELK4的表达有任何显著的增加。然而,根据最近研究得知ELK1和ELK4 结合motif富集在O-GlcNAc标记的染色质中,在选定的耐药相关基因中确认O-GlcNAc有对应H3K4me3标记。OGT是唯一能够催化单环糖O-GlcNAc添加到丝氨酸和苏氨酸残基上,改变其功能、稳定性和定位的蛋白质。重要的是,有报道称OGT可以调控SET1/COMPASS介导的H3K4me3重塑(Deplus, Delatte et al.2013)。此外,OGT还与TET1蛋白高度相关,以调节胚胎干细胞中的H3K4me3标记。OGT和TET1与H3K4me3peak共定位于未甲基化且富含CpG的启动子。因此,作者推测OGT和TET1可能参与了H3K4me3的重塑过程。
在IDTC colony状态下,通过WB检测OGT和TET1蛋白水平上调(图. 4a)。因此,在IDTC colony状态下O-GlcNAc糖基化水平的增加证实了OGT催化活性的增加(图. 4b)。相反,早期 IDTC状态下,OGT和TET1的表达并没有一致增加(图.4a),这说明这些酶的特异性表达与H3K4me3水平相一致(图.2a)。使用先前报道的黑色素瘤患者使用MEK抑制剂治疗30天后的异种移植模型TAK-733与IDTC colony状态样本进行WB检测表明在大多数样本中显示OGT、TET1和O-GlcNAc糖基化增加,且与基因突变背景无关。
据报道,OGT活性由己糖胺生物合成途径(HBP)提供原料。对WM164 parent、IDTC、IDTC colony和90天处理后耐药细胞进行代谢组分析,结果表明HBP在IDTC colony状态下显著激活,其终产物UDP-N -乙酰氨基葡萄糖等的水平增加(图.4d、4e)。UDP - N -乙酰氨基葡萄糖是OGT催化O-GLCN酰化反应的底物。
此外,免疫沉淀发现OGT的使得组蛋白H3的O-GlcNAcy增加,表明在IDTC colony状态下OGT介导的表观遗传修饰增加。值得注意的是,尽管药物诱导的O-GlcNAc糖基化水平在所有细胞系中都有明显表达,但H3的O-GlcNAc糖基化水平明显增加(图. 4f)。OSMI4B可抑制OGT,这是一种最近开发的用于体外研究的细胞穿透性OGT抑制剂,在IDTC colony状态有效下调H3K4me3水平,证实了OGT在H3K4me3重塑中的作用(图.4g)。抑制OGT还下调了IDTC colony状态下BRAF抑制剂耐药相关基因NRG1、WNT5A、VEGFC和EGFR的表达,且这些基因也与H3K4me3重塑相关(图.4h)。此外,在H3K4me3 ChIP-seq数据中显示motif富集的转录因子FLI1和ARNT的表达水平也显示出OGT的依赖性,这表明OGT对H3K4me3重塑有直接或者间接的影响。这些数据表明,在IDTC菌落状态下,OGT在H3K4me3重塑和获得性耐药表型中发挥重要作用。
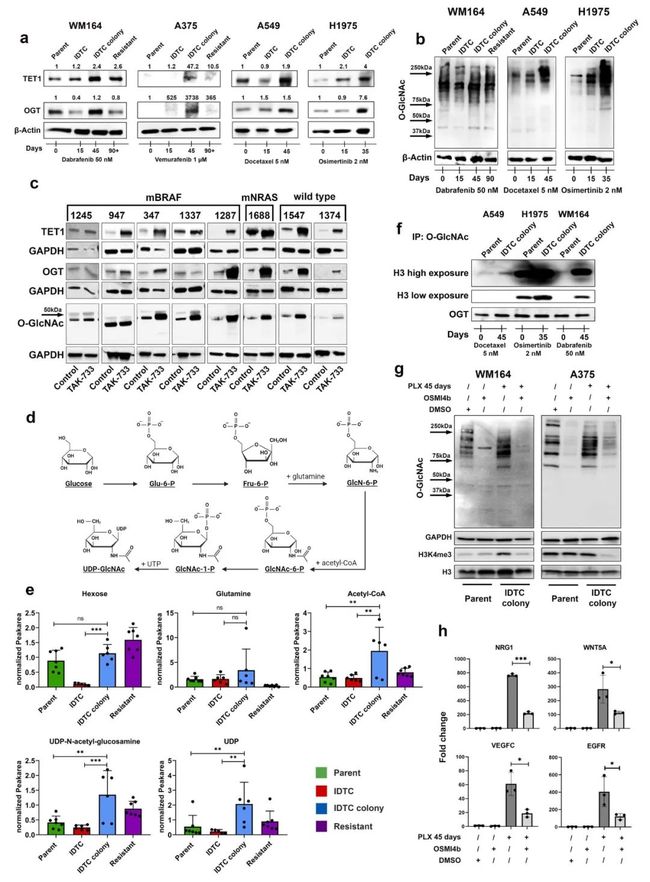
图4 早期耐药IDTC细胞通过O-linked n -乙酰氨基葡萄糖转移酶上调H3K4me3
5、抑制OGT阻断治疗诱导的细胞重编程,预防肿瘤复发
确定OGT与耐药相关基因及H3K4me3重新编程后,构建shRNA干扰的OGT敲除细胞株(图.5a)。在体外,OGT的下调不会降低细胞的生长速率,也不会使短期给药的细胞变得敏感。然而,长期药物暴露后,与shControl细胞相比,shOGT细胞形成IDTC colony的能力明显下降(图.5a)。TET1的抑制并没有持续影响生长速度或药物敏感性,但降低了用药处理后IDTC colony形成的效率(图.5b)。长期暴露于BRAF抑制剂的shOGT细胞群中形成的稀疏IDTC colony表明,OGT转录水平被重新激活,这突出了OGT对这种表型形成的重要性。
与shControl细胞相比,长期用药的shOGT和shTET1细胞中H3K4me3重获,OGT和TET1表达增加。与shControl相比,shOGT和shTET1细胞在体内生长没有差异,但对BRAF/MEK抑制的反应延长,导致与shControl小鼠相比,肿瘤负荷显著降低(图.5c)。WB结果显示,shControl组OGT、TET1和OGlcNAc水平均升高,而shOGT组OGT、TET1和OGlcNAc水平降低(图.5d)。免疫荧光显示,与shControl相比,BRAF/MEK抑制剂治疗的shOGT和shTET1肿瘤的H3K4me3水平仍然较低(图.5e)。OGT在体内的重要性也在肺癌异种移植模型中得到了证实,该模型使用KRAS突变体A549细胞,每周腹腔注射多西他赛(图.5f)。总体来说,OGT和TET1通过使癌细胞重新获得增殖能力,在获得性耐药的出现中发挥关键作用。
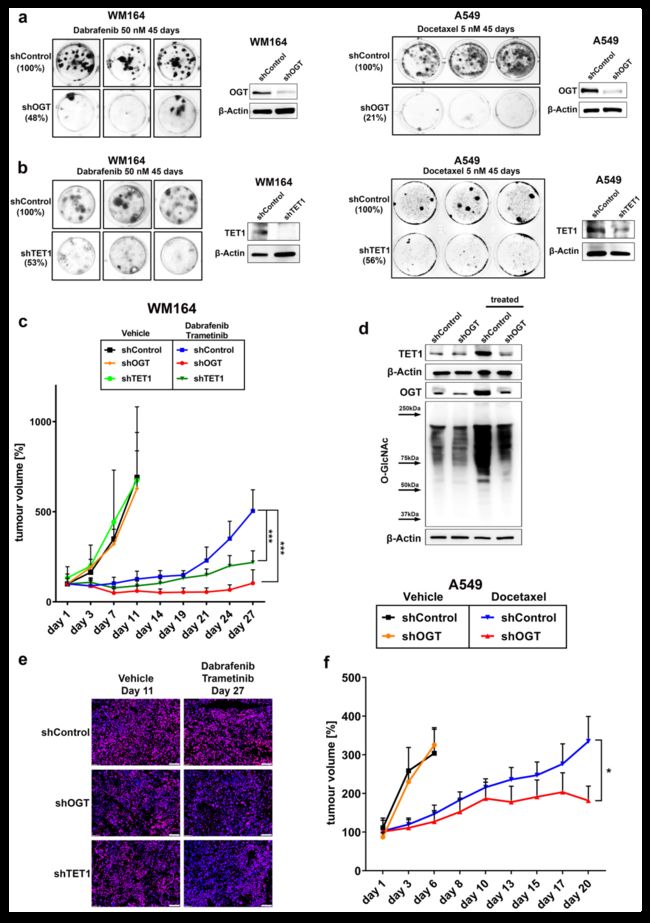
图5 抑制OGT阻断耐药细胞重编
6、AMP活化的蛋白激酶(AMPK)激活阻止OGT介导的细胞重编程
由于体内使用的特异性OGT抑制剂的开发尚未成功,作者决定探索抑制OGT功能的间接途径。有报道表明,AMPK能够通过降低HBP通量来阻止O-GlcNAc糖基化(Gelinas et al. 2018),并以mtor依赖的方式直接调控OGT。因此,抗癌药物的组合与单次治疗相比,使用AMPK激活剂时,阿司匹林可防止IDTC colony形成(图.6a)。此外,达拉菲尼与阿司匹林或另一种AMPK激活剂二甲双胍联合使用,抑制了WM164黑色素瘤细胞增殖的再激活,通常在45-80天之间(图.6b)。达拉菲尼联合阿司匹林治疗WM164细胞235天,21天停药后后恢复药敏(图.6c)。阿司匹林联合治疗45天可诱导AMPK靶蛋白乙酰辅酶a羧化酶(p-ACC)的磷酸化,抑制药物诱导的OGT和O-GlcNAcylation的增加(图6d)以及H3K4me3的重新激活(图6e)。在阿司匹林联合不同化疗药物长期治疗后,多个细胞系中OGT转录水平也被发现下降,说明AMPK活化可作为不错的选择干扰治疗诱导的OGT表达。干扰RNA介导的AMPKα1/2催化亚基(PRKAA1/PRKAA2)基因敲除消除了阿司匹林介导的H3K4me3下调,即使在阿司匹林存在的情况下也能导致IDTC colony的再现(图.6f)。
通过黑色素瘤异种移植,评估BRAF/MEK抑制剂与阿司匹林的体内联合治疗效果。与体外实验结果一致,阿司匹林单独使用对肿瘤生长没有影响,但与达拉菲尼/曲美替尼联合使用相比,达拉菲尼/曲美替尼联合使用对肿瘤的反应较长,治疗约19-21天后开始复发,导致实验结束时肿瘤体积有显著差异(图.6g)。WB 和免疫荧光染色显示OGT、TET1和H3K4me3随处理时间变化,与在达拉菲尼/曲美替尼/阿司匹林三联疗法治疗肿瘤时发现的OGT、O-GlcNAc和H3K4me3减少的结果一致(图6h和6i)。提示阿司匹林可以在体内阻断治疗诱导的代谢和表观遗传重编程。
作者检测了WM164细胞在IDTC集落状态下的产生肿瘤潜能以及长期联合阿司匹林治疗的效果。与未处理的亲代细胞相比,暴露于达拉菲尼(IDTC colony state) 50天的WM164细胞体内生长潜力显著增加,阿司匹林联合治疗可防止这一现象出现。从肿瘤中分离出来的细胞经药物治疗后,在体外恢复了药物敏感性,表明短暂的表观遗传重编程足以诱导疾病复发。总之,作者确定AMPK作为目标靶点,可以干扰OGT介导的获得性耐药的发展进程。

图6 AMP活化蛋白激酶(AMPK)的激活阻止OGT介导的细胞重编程
研究结论
本文先从细胞层面探究肿瘤耐药形成机制,再至动物造模验证。H3K4me3重塑广泛存在于CpG岛区域和与OGlcNAc标记的染色质相关的DNA结合基序(motif)中。OGT和TET1的下调抑制了体外IDTC colony的形成,延缓了体内获得性耐药。这种表观遗传适应过程也可以通过激活AMPK来抑制OGT的表达和活性而间接被阻断。值得注意的是整个文章的方向是多元化考量,并不是数据分析出来的相关基因一直往下延续,看似结果无方向(其实是文献积累)更要多读文献多条道路找找方向。