双侧条形图绘制教程
写在前面
双侧条形图在我们的文章中也是比较常见的,那么这样的图形是如何绘制的呢? 以及它使用的数据类型是什么呢? 这些都是我们在绘制图形前需要掌握的,至少我们知道绘图的数据集如何准备,这样才踏出第一步。
今天的教程,我们会从数据的准备,以及数据如何整理,以及结合自己的绘图过程中遇到问题是如何解决来进行讲解。PS:仅代表个人的观点,以及自己遇到此问题时自己的方法来进行说明。也许,这个并不会死唯一且最好的方法,大家在绘图中请结合自己的能力和方法。
本期教程

获得本期教程
代码口令:20240205
原文链接:R语言绘图教程 | 双侧条形图绘制教程
什么时间及什么数据使用双侧条形图?一般使用此图形,基本是富集图
一般使用此图形,基本是富集图。如GO、KEGG、GSEA、GSVE等等,我们用来表示各个通路在研究中的显著性、得分情况等。
绘图
教程一
这里我们使用已经整理好的数据进行绘图,我们使用Execl进行整理数据。数据结果来源,GO、KEGG、GSEA、GSVE等富集结果。
1. 导入所需的R包
##'@导入所需的R包
library(limma)
library(ggplot2)
library(ggpubr)
library(tidyr)
library(tidyverse)
library(ComplexHeatmap)
2. 导入数据
在这里我们发现,我们有很多个富集通路,但是我们绘图的时候需要这么多吗?应该只需要各别几个。在这里我们可以手动调整,或是通过P值进行筛选。
dt1 <- read.csv("DE_KEGG.input.csv",header = T, row.names = 1)
###'@导入txt文件
#dt1 <- read.table("Input_KEGG.txt",header = T, row.names = 1, sep = '\t',check.names = F)
> dt1[1:5,1:5]
logFC AveExpr t P.Value adj.P.Val
KEGG_TGF_BETA_SIGNALING_PATHWAY -0.1723618 0.020400811 -5.029813 0.000001250 0.000190767
KEGG_COLORECTAL_CANCER -0.1443645 0.007857589 -3.773441 0.000222656 0.010260593
KEGG_MELANOMA -0.1129672 0.041504860 -3.736434 0.000255165 0.010260593
KEGG_ADHERENS_JUNCTION -0.1541395 0.027103539 -3.721243 0.000269769 0.010260593
KEGG_PATHWAYS_IN_CANCER -0.1100711 0.017776668 -3.615438 0.000395646 0.010260593
3. 筛选数据
筛选出的作图的数据,这里我们的直接使用Description,LogP和group进行绘图
df <- data.frame(Description = rownames(dt1), LogP = log10(dt1$adj.P.Val), Group = dt1$group, LogFC = dt1$logFC)

4. 调整Description顺序
###'@调整`Description`顺序
df$Description <- factor(df$Description, levels = rev(df$Description))
若是我们这里有自己整理的Description顺序,可以直接在levels()后面加上自己的排序即可。
绘图
- 基础图形
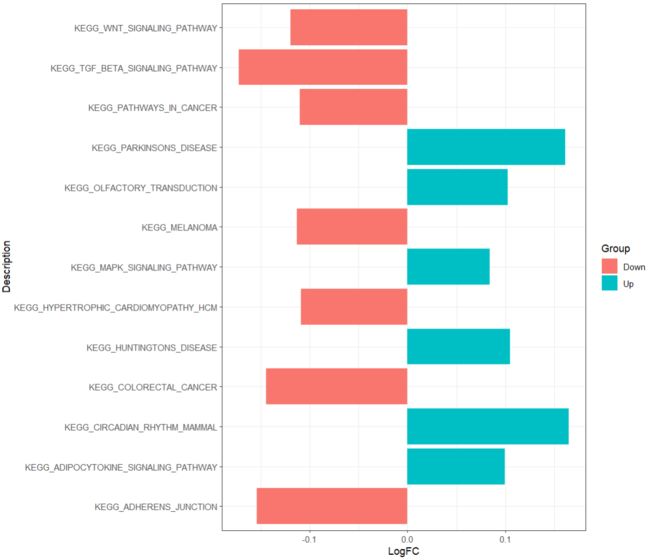
ggplot(df, aes(x = LogFC, y = Description, fill = Group))+
geom_col()+
theme_bw()
2.更改颜色
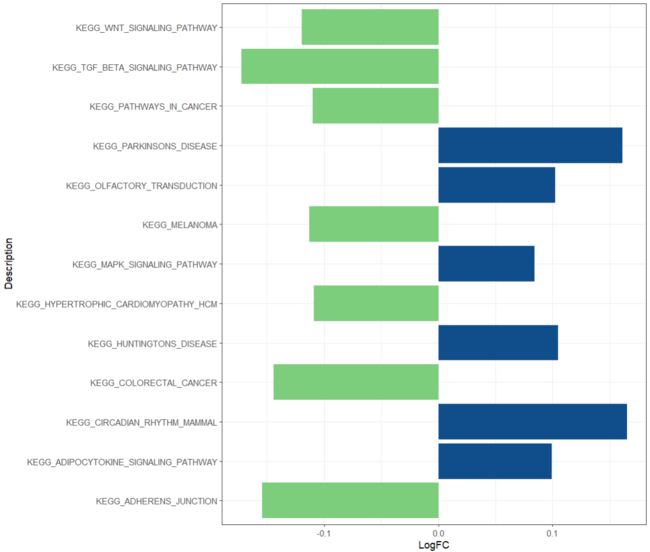
ggplot(df, aes(x = LogFC, y = Description, fill = Group))+
scale_fill_manual(values = c('palegreen3','dodgerblue4'), guide = FALSE)+
geom_col()+
theme_bw()
- 高阶绘图
此教程来源:https://mp.weixin.qq.com/s/aVy5ubaKc6Q8_wf-R9TVGQ
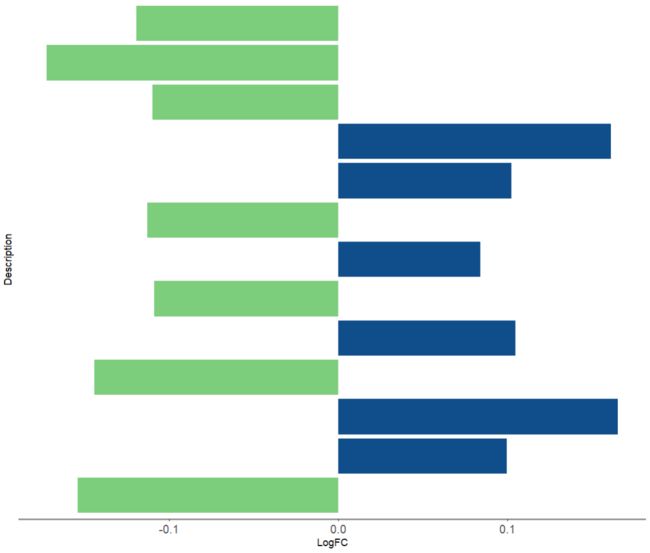
p1 <- ggplot(df, aes(x = LogFC, y = Description, fill = Group))+
scale_fill_manual(values = c('palegreen3','dodgerblue4'), guide = FALSE)+
geom_col()+
theme_bw()+
##'@自定义主题
theme(
legend.position = 'none',
axis.text.y = element_blank(),
axis.ticks.y = element_blank(),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.border = element_blank(),
axis.line.x = element_line(color = 'grey60',size = 1.1),
axis.text = element_text(size = 12)
)
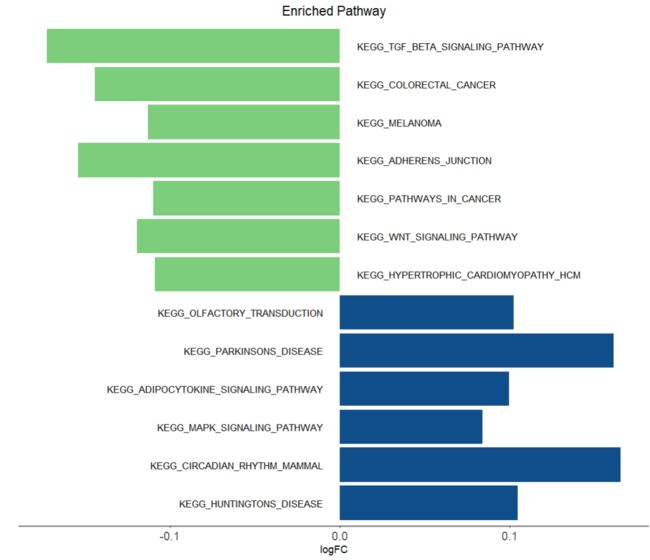
添加对应的通路标签
up <- df[which(df$Group == 'Up'),]
down <- df[which(df$Group == 'Down'),]
p1 +
##'@添加Up的通路富集
geom_text(data = up,
aes(x = -0.01, ## 调整通路标签x轴位置
y = Description, label = Description),
size = 3.5,
hjust = 1)+ #标签右对齐
##'@添加Down的通路标签
geom_text(data = down,
aes(x = 0.01, y = Description, label = Description),
size = 3.5,
hjust = 0)+ #标签左对齐
#scale_x_continuous(breaks=seq(-4, 6, 2)) + #x轴刻度修改,若需要请结合自己的数据调整
labs(x = 'logFC', y = ' ', title = 'Enriched Pathway') + #修改x/y轴标签、标题添加
theme(plot.title = element_text(hjust = 0.5, size = 14))
教程二
若是我们的直接导入通路数据集矩阵,以及我们的未有logFC和P值,我们该如何操作呢?
第一步需要操作步骤:进行差异分析。比如,我们输出数据是GSVA的数据集。
- 输入数据集
data_df <- read.csv("Input_KEGG.csv",header = T,row.names = 1)
##'@查看数据
data_df[1:5,1:10]
> data_df[1:5,1:10]
sample001 sample002 sample003
KEGG_O_GLYCAN_BIOSYNTHESIS -0.04304991 -0.24987132 -0.12401679
KEGG_AMINO_SUGAR_AND_NUCLEOTIDE_SUGAR_METABOLISM -0.24783431 -0.11383994 -0.47895615
KEGG_GLYCOSAMINOGLYCAN_DEGRADATION 0.26018439 0.01315923 -0.25700397
KEGG_GLYCOSAMINOGLYCAN_BIOSYNTHESIS_CHONDROITIN_SULFATE 0.42760250 -0.06222532 0.00031469
KEGG_GLYCOSAMINOGLYCAN_BIOSYNTHESIS_KERATAN_SULFATE 0.34888990 -0.38251570 -0.26030188
sample004 sample005 sample006
KEGG_O_GLYCAN_BIOSYNTHESIS -0.11976645 0.40799471 -0.121623478
KEGG_AMINO_SUGAR_AND_NUCLEOTIDE_SUGAR_METABOLISM -0.52727188 0.31490335 -0.000949728
KEGG_GLYCOSAMINOGLYCAN_DEGRADATION -0.03605253 0.29166666 0.001121143
KEGG_GLYCOSAMINOGLYCAN_BIOSYNTHESIS_CHONDROITIN_SULFATE 0.09261353 -0.06208377 -0.159061239
KEGG_GLYCOSAMINOGLYCAN_BIOSYNTHESIS_KERATAN_SULFATE -0.43428522 0.15120498 -0.455994453
sample007 sample008 sample009
- 差异分析
在group_list设置中,请根据自己数据进行设置,比如我这里有160个样本,CK和Treat各80个。
###'@构建design
group_list <- data.frame(sample = colnames(data_df), group = c(rep("CK", 80), rep("Treat", 80)))
group_list$group <- factor(group_list$group,levels=c("CK",'Treat'))
design <- model.matrix(~ 0 + group_list$group)
colnames(design) <- levels(group_list$group)
rownames(design) <- colnames(data_df)
design
- 构建差异比较矩阵
###'@构建差异比较矩阵
contrast.matrix <- makeContrasts(CK-Treat, levels = design)
- 差异分析
fit <- lmFit(data_df, design)
fit2 <- contrasts.fit(fit, contrast.matrix)
fit2 <- eBayes(fit2)
- 获得差异结果
dt <- topTable(fit2, coef = 1, n = Inf, adjust.method = "BH", sort.by = "P")
dt[1:10,1:5]
> dt[1:10,1:5]
logFC AveExpr t P.Value adj.P.Val
KEGG_TGF_BETA_SIGNALING_PATHWAY -0.17236176 0.020400811 -5.029813 1.246843e-06 0.000190767
KEGG_COLORECTAL_CANCER -0.14436446 0.007857589 -3.773441 2.226562e-04 0.010260593
KEGG_MELANOMA -0.11296720 0.041504860 -3.736434 2.551651e-04 0.010260593
KEGG_ADHERENS_JUNCTION -0.15413947 0.027103539 -3.721243 2.697688e-04 0.010260593
KEGG_PATHWAYS_IN_CANCER -0.11007105 0.017776668 -3.615438 3.956464e-04 0.010260593
KEGG_WNT_SIGNALING_PATHWAY -0.11940334 0.014475730 -3.610726 4.023762e-04 0.010260593
KEGG_HYPERTROPHIC_CARDIOMYOPATHY_HCM -0.10903295 0.035562175 -3.462058 6.792812e-04 0.014847145
KEGG_GAP_JUNCTION -0.10486217 0.006571587 -3.358319 9.693892e-04 0.014898594
KEGG_LONG_TERM_POTENTIATION -0.09738506 0.003833619 -3.309357 1.143329e-03 0.014898594
KEGG_SPHINGOLIPID_METABOLISM -0.12157635 0.031751704 -3.306694 1.153577e-03 0.014898594
#把通路的limma分析结果保存到文件
write.table(dt, "all.limma_KEGG.output.txt", quote = F,sep = '\t',row.names = T,col.names = T)
#write.csv(x,"all.limma_KEGG.output.csv")
到这里我们也就可以使用以上的代码进行分析了,你可以使用P值或是LogFC进行绘制,我们论文中一般使用的是P值进行绘制图图形。
在这里,我们使用t值绘制图形。操作如下所示。
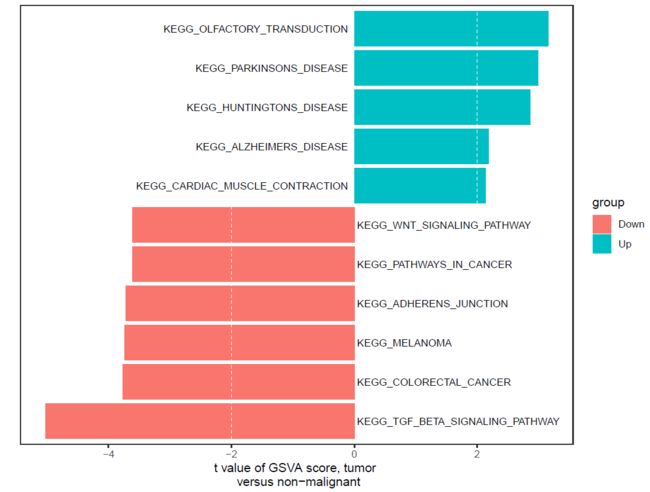
- 筛选所需数据,使用t值进行过滤,t值的大小根据自己的需求进行调整
df_t <- df_t[df_t$score > 2 | df_t$score < -3.5,]
dim(df_t)
df_t
> df_t
Description score group
1 KEGG_TGF_BETA_SIGNALING_PATHWAY -5.029813 Down
2 KEGG_COLORECTAL_CANCER -3.773441 Down
3 KEGG_MELANOMA -3.736434 Down
4 KEGG_ADHERENS_JUNCTION -3.721243 Down
5 KEGG_PATHWAYS_IN_CANCER -3.615438 Down
6 KEGG_WNT_SIGNALING_PATHWAY -3.610726 Down
18 KEGG_OLFACTORY_TRANSDUCTION 3.157596 Up
26 KEGG_PARKINSONS_DISEASE 2.990963 Up
31 KEGG_HUNTINGTONS_DISEASE 2.863284 Up
61 KEGG_ALZHEIMERS_DISEASE 2.182530 Up
64 KEGG_CARDIAC_MUSCLE_CONTRACTION 2.133893 Up
- 使用t值进行分组
cutoff <- 2
df_t$group <- case_when(df_t$score > cutoff ~ "Up", df_t$score < cutoff ~ "Down")
df_t[1:9,1:3]
> df_t[1:9,1:3]
Description score group
1 KEGG_TGF_BETA_SIGNALING_PATHWAY -5.029813 Down
2 KEGG_COLORECTAL_CANCER -3.773441 Down
3 KEGG_MELANOMA -3.736434 Down
4 KEGG_ADHERENS_JUNCTION -3.721243 Down
5 KEGG_PATHWAYS_IN_CANCER -3.615438 Down
6 KEGG_WNT_SIGNALING_PATHWAY -3.610726 Down
18 KEGG_OLFACTORY_TRANSDUCTION 3.157596 Up
26 KEGG_PARKINSONS_DISEASE 2.990963 Up
31 KEGG_HUNTINGTONS_DISEASE 2.863284 Up
- 进行排序
df_sort <- df_t[order(df_t$score),]
df_sort$Description <- factor(df_sort$Description, levels = df_sort$Description)
df_sort[1:9,1:3]
- 绘图
ggplot(df_sort, aes(Description, score, fill = group)) + geom_bar(stat = 'identity') +
coord_flip() +
#scale_fill_manual(values = c('palegreen3','dodgerblue4'), guide = FALSE) +
#画2条虚线
geom_hline(yintercept = c(-cutoff,cutoff),
color="white",
linetype = 2, #画虚线::1表示实线,2表示虚线,3表示间距更小的虚线
size = 0.3)+ #线的粗细
geom_text(data = subset(df_sort, score < 0),
aes(x=Description, y= 0, label= paste0(" ", Description), color = group),#bar跟坐标轴间留出间隙
size = 3, #字的大小
hjust = "bottom" ) + #字的对齐方式
geom_text(data = subset(df_sort, score > 0),
aes(x=Description, y= -0.1, label=Description, color = group),
size = 3, hjust = "inward",angle=360) +
scale_colour_manual(values = c("black","black"), guide = FALSE)+
xlab("") +ylab("t value of GSVA score, tumor \n versus non-malignant")+
theme_bw() + #去除背景色
theme(panel.grid =element_blank()) + #去除网格线
theme(panel.border = element_rect(size = 0.6)) + #边框粗细
theme(axis.line.y = element_blank(), axis.ticks.y = element_blank(),
axis.text.y = element_blank()) #去除y轴
代码和数据链接:
本教程涉及的数据、代码和文件等在社群中可获得!!
若我们的分享对你有用,希望您可以点赞+收藏+转发,这是对小杜最大的支持。
往期文章:
1. 复现SCI文章系列专栏
2. 《生信知识库订阅须知》,同步更新,易于搜索与管理。
3. 最全WGCNA教程(替换数据即可出全部结果与图形)
-
WGCNA分析 | 全流程分析代码 | 代码一
-
WGCNA分析 | 全流程分析代码 | 代码二
-
WGCNA分析 | 全流程代码分享 | 代码三
-
WGCNA分析 | 全流程分析代码 | 代码四
-
WGCNA分析 | 全流程分析代码 | 代码五(最新版本)
4. 精美图形绘制教程
- 精美图形绘制教程
5. 转录组分析教程
转录组上游分析教程[零基础]
一个转录组上游分析流程 | Hisat2-Stringtie
小杜的生信筆記 ,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!