python小白入门单细胞分析scanpy
大家好,今天我们分享scanpy的标准流程
基本概念介绍
Scanpy 和 Seurat 基本上完全一样,Scanpy 构建的对象叫做 AnnData 对象,他的数据存储是以4 个模块存储(如下图)
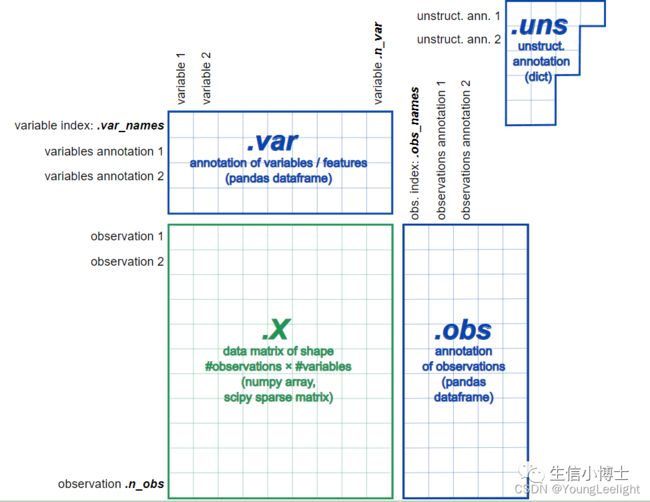
如果你不理解scanpy这种数据结构的话,可以对比学习一下seurat中数据结构 单细胞直播三seurat数据结构与数据可视化
其中X对象为count 矩阵。这里要注意一下,它和 R 语言的不同,Scanpy 中的行为样本,列为基因。这也和 python 的使用习惯相关
-
obs 存储的是 seurat 对象中的 meta.data 矩阵
-
X 对象为count 矩阵,与 seurat 对象是转置关系
-
var 存储的是基因(特征)的信息
-
uns 存储的是后续添加的非结构信息
官方示例代码
import scanpy as sc
import os
import math
import itertools
import warnings
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
%matplotlib inline
%config InlineBackend.figure_format = 'svg'
warnings.filterwarnings("ignore")
plt.rc('font',family='Times New Roman')
my_colors = ["#1EB2A6","#ffc4a3","#e2979c","#F67575"]
sc.settings.verbosity = 3 # 输出提示信息
# ?sc.settings.verbosity
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80, facecolor='white')# 设置输出图像格式
results_file = 'write/pbmc3k.h5ad' # 存储分析结果
scanpy==1.6.0 anndata==0.7.5 umap==0.4.6 numpy==1.19.2 scipy==1.4.1 pandas==1.1.3 scikit-learn==0.23.2 statsmodels==0.12.0这里的读取文件的方式和R语言构造seurat对象基本一样 (按照官网分类有12中读取方式)
下面主要介绍两种方法
第一种方法是,文件下面要有3个初始文件包括:
-
barcord
-
genes
-
matrix
然后使用输 sc.read_10_mtx 读取
第二种方法是直接构建AnnData对象
然后分别的将表达矩阵,细胞信息,基因信息读取,代码如下
# 这个是第二种方法
# creat scanpy object
#df = pd.read_csv('processfile/count.csv', index_col=0)
#meta = pd.read_csv('processfile/metadata.csv', index_col=0)
#cellinfo = pd.DataFrame(df.index,index=df.index,columns=['sample_index'])
#geneinfo = pd.DataFrame(df.columns,index=df.columns,columns=['genes_index'])
#sce = sc.AnnData(df, obs=cellinfo, var = geneinfo)
# 这个是第一种读取方法
adata = sc.read_10x_mtx(
'./filtered_gene_bc_matrices/hg19/', # the directory with the `.mtx` file
var_names='gene_symbols', # use gene symbols for the variable names (variables-axis index)
cache=True)
adata.var_names_make_unique()
adatatips: pytho和R语言有点不同,通常情况下,行为样本, 列为特征
adata.obs.shape # 2700个细胞
adata.var.shape # 32738个基因
adata.to_df().shape # 2700*32738
adata.obs.head()
adata.var.head()
adata.to_df().iloc[0:5,0:5]数据预处理
这里介绍一下scanpy中常用的组件
-
pp: 数据预处理
-
tl: 添加额外信息
-
pl:可视化
统计基因在细胞中的占比并可视化
sc.pl.highest_expr_genes(adata, n_top=20) # 每一个基因在所有细胞中的平均表达量(这里计算了百分比含量)
sc.pp.filter_cells(adata, min_genes=200) # 每一个细胞至少表达200个基因
sc.pp.filter_genes(adata, min_cells=3) # 每一个基因至少在3个细胞中表达
过滤线粒体DNA
str.startswith 不支持正则,如果要使用正则则使用.str.match
sce.var_names[sce.var_names.str.match(r'^MT-')]
sce.var_names[sce.var_names.str.match(r'^RP[SL0-9]')]
sce.var_names[sce.var_names.str.match(r'^ERCC-')]
# 抽取带有MT的字符串
adata.var['mt'] = adata.var_names.str.startswith('MT-')
# 数据过滤
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
# 过滤后可视化(官方文档真的骚到我头皮发麻)
sc.pl.violin(adata, ['n_genes_by_counts'],jitter=0.4)
sc.pl.violin(adata, ['total_counts'],jitter=0.4)
sc.pl.violin(adata, ['pct_counts_mt'],jitter=0.4)sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt')
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts')# 提取线粒体dna在5%以下
adata = adata[adata.obs.pct_counts_mt < 5, :]
# 提取基因不超过2500的细胞
adata = adata[adata.obs.n_genes_by_counts < 2500, :]下面就是scanpy的标准流程:
-
log : NormalizeData
-
找特征 : FindVariableFeatures
-
标准化 : ScaleData
-
pca : RunPCA
-
构建图 : FindNeighbors
-
聚类 : FindClusters
-
tsne /umap : RunTSNE RunUMAP
-
差异基因 : FindAllMarkers / FindMarkers
sc.pp.normalize_total(adata, target_sum=1e4) # 不要和log顺序搞反了 ,这个是去文库的
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
# 可视化
sc.pl.highly_variable_genes(adata)
# 保存一下原始数据
adata.raw = adata# 提取高变基因
adata = adata[:, adata.var.highly_variable]
# 过滤掉没用的东西
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
# 中心化
sc.pp.scale(adata, max_value=10)
# pca
sc.tl.pca(adata, svd_solver='arpack')
sc.pl.pca(adata, color='CST3')
sc.pl.pca_variance_ratio(adata, log=True)
# 输出结果
adata.write(results_file)# 构建图
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
sc.tl.umap(adata)
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'])
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'], use_raw=False)
sc.tl.tsne(adata)
sc.pl.tsne(adata, color=['CST3', 'NKG7', 'PPBP'])
sc.pl.tsne(adata, color=['CST3', 'NKG7', 'PPBP'], use_raw=False)

sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
sc.tl.leiden(adata)
sc.pl.umap(adata, color=['leiden', 'CST3', 'NKG7'])
sc.pl.tsne(adata, color=['leiden', 'CST3', 'NKG7'])
# 保存结果
adata.write(results_file)

找差异基因
# 这里使用秩和检验
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
adata.write(results_file)
num = 2 # 通过这个控制marker基因的数量
marker_genes = list(set(np.array(pd.DataFrame(adata.uns['rank_genes_groups']['names']).head(num)).reshape(-1)))
len(marker_genes)
# 看一下每一个组的特征基因
adata = sc.read(results_file)
result = adata.uns['rank_genes_groups']
groups = result['names'].dtype.names
pd.DataFrame(
{group + '_' + key[:1]: result[key][group]
for group in groups for key in ['names', 'pvals']}).iloc[0:6,0:6]
# 比较组别间差异
sc.tl.rank_genes_groups(adata, 'leiden', groups=['0'], reference='1', method='wilcoxon')
sc.pl.rank_genes_groups(adata, groups=['0'], n_genes=20)
sc.pl.rank_genes_groups_violin(adata, groups='0', n_genes=8)
# 这里需要重载一下结果,如果不重载的话结果会有差异的
adata = sc.read(results_file)
sc.pl.rank_genes_groups_violin(adata, groups='0', n_genes=8)
sc.pl.violin(adata, ['CST3', 'NKG7', 'PPBP'], groupby='leiden')

new_cluster_names = [
'CD4 T', 'CD14 Monocytes',
'B', 'CD8 T',
'NK', 'FCGR3A Monocytes',
'Dendritic', 'Megakaryocytes']
adata.rename_categories('leiden', new_cluster_names)
sc.pl.umap(adata, color='leiden', legend_loc='on data', title='', frameon=False, save='.pdf')
sc.pl.dotplot(adata, marker_genes, groupby='leiden');
sc.pl.stacked_violin(adata, marker_genes, groupby='leiden', rotation=90);
adata.raw.to_adata().write('./write/pbmc3k_withoutX.h5ad')
WARNING: saving figure to file figures\umap.pdf

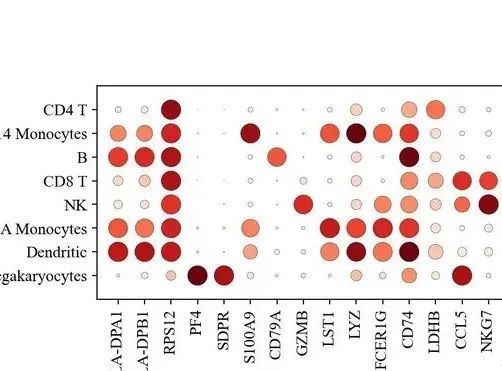
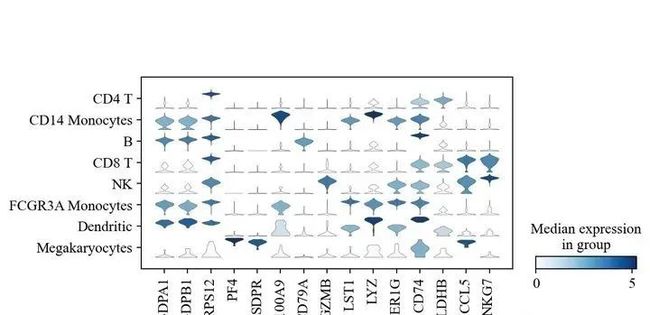
如果你能看到这里
那我再给你看下我是如何使用scanpy处理一个covid-19超大数据集的
GSE158055 covid19 肺组织60W单细胞细胞实战
应粉丝呼声,欢迎假如我们的生信技能互换群

更多精彩关注公众号:生信小博士

![]()
看完记得顺手点个“在看”哦!