scIMC:scRNA-seq插补方法基准
在scRNA-seq中一个主要的挑战即为“dropout”事件,它扭曲了基因表达,显著影响了单细胞转录组的下游分析。为了解决这个问题,已经做了很多努力,并开发了几种基于模型和基于深度学习的scRNA-seq插补方法。但是,目前还缺乏对现有方法进行全面、系统的比较。在这项工作中,作者使用6个模拟和2个真实的scRNA-seq数据集,从以下四个方面全面评估和比较了总共12种可用的插补方法:1.基因表达恢复,2.细胞聚类,3.基因差异表达,4.细胞轨迹重建。最终得到结论,在主要的基准比较下,基于深度学习的方法通常比基于模型的方法表现出更好的整体性能,这表明深度学习在插补方面的力量。
来自:scIMC: a platform for benchmarking comparison and visualization analysis of scRNA-seq data imputation methods
网站:https://server.wei-group.net/scIMC/
目录
- 背景概述
-
- 基于模型的方法
- 基于深度学习的方法
- 使用方法
-
- 基准数据集
- 评价指标
- 额外的工具
- 结果
-
- 基因差异表达分析
- 轨迹分析
背景概述
据估计,人体共有 4 × 1 0 13 4×10^{13} 4×1013个细胞,它们表现出不同的形态和功能。细胞转录组分析在表征细胞生物学状态方面起着重要作用。传统的bulk RNA测序(bulk RNA-seq)技术可以检测细胞群体的平均基因表达水平。然而,基因在不同细胞中的表达水平存在差异。因此,这种技术无法量化细胞间的异质性。scRNA-seq(scRNA-seq)技术的最新发展使研究人员能够研究不同细胞间基因表达的细胞间异质性,发现新的细胞类型,提高细胞分辨率水平。
然而,scRNA-seq也带来了新的计算挑战。主要的挑战是目前的技术缺陷,如低RNA捕获和测序效率,导致无法检测到表达的基因,很大比例的表达基因为零或低读取计数。观察到的零值并不能反映真实的基因表达,这被定义为“dropout”事件。“dropout”事件引入了很高的技术噪声,使分析scRNA-seq数据变得困难。然而,并非scRNA-seq数据中的所有零都可以视为“dropout”事件。因为存在真正的零表达事件,代表特定细胞类型中的低水平基因表达。因此,在scRNA-seq数据中区分“假”(dropout)和“真”(biological gene silencing)的零计数是具有挑战性的。因此,迫切需要处理“dropout”事件。
插补是恢复“dropout”事件的常用方法。在最近的研究中提出了几种scRNA-seq数据插补方法。作者将现有的方法大致分为两类:1.基于模型的方法和2.基于深度学习的方法。
基于模型的方法
利用细胞间的先验知识和信息预测缺失值的基于模型的插补方法,包括利用细胞-细胞相互作用网络、基因-基因相互作用网络以及两者的整合进行插值。这些方法根据相似的细胞信息、相似的基因信息和使用先验知识的数学计算来恢复目标基因的表达。例如,SAVER(single cell Analysis Via Expression Recovery)是一种优化整个基因表达计数的方法,利用基因和细胞之间的信息来提高所有基因的表达值。同样,MAGIC(Markov Affinity-based Graph Imputation of Cells)应用数据扩散在相似的细胞之间共享信息,以优化基因表达矩阵,并计算缺失值。MAGIC和SAVER都优化了所有基因表达,包括那些不受“dropout”事件影响的基因表达,同时增加了将新噪声引入其余数据的可能性。为了解决这个问题,提出了一种名为scImpute的新方法,该方法首先自动识别可能的“dropout”事件,然后执行imputation,以避免向其余数据引入新的噪声。后来又提出了一种利用聚类结果识别多组相似细胞,并通过平均相似细胞的表达值进行imputation的方法DrImpute。
随着相互作用网络的出现,合理使用先验网络已成为插补方法的关键。scNPF在相似的细胞之间共享信息,并使用相互作用网络的先验知识来确定给定细胞的基因表达。scNPF的优势在于它不仅可以利用生物网络中存储的丰富结构,还可以捕获上下文特定的信息来增强基因之间的关系。除scNPF外,netNMF-sc还使用网络正则化非负矩阵分解NMF将计数矩阵分解为两个低维矩阵:基因embedding矩阵和细胞embedding矩阵。网络正则化使网络中连接的两个基因在低维基因矩阵中具有相似的表示,从而恢复数据结构。
基于深度学习的方法
近年来,深度学习已广泛应用于scRNA-seq领域,如DeepMAPS用于生物网络推理。基于深度学习的方法可以捕捉基因表达的隐藏分布,并学习基因表达分布模型的参数来估算缺失值。例如,AutoImpute是一种基于自编码器和稀疏基因表达矩阵的插补方法。它可以学习输入数据的固有分布,并估计缺失值。Lopea等人开发了单细胞变分推理scVI,利用随机优化和深度神经网络来聚合相似细胞和基因之间的信息,并估计计数矩阵的基本分布。然而,上述方法不能有意保留生物零点(真零计数),也不能扩展到分析数千个细胞的大型数据集。为了解决这个问题,提出了一种自适应阈值低秩近似(ALRA)方法。它能够通过非负性和相关性结构选择性地输入缺失值,并在输入“dropout”事件的同时有效地保持生物零点。深度计数自动编码器网络(DCA)通过引入具有零膨胀的负二项式噪声模型来捕捉非线性基因-基因相关性,同时考虑数据的计数分布、稀疏性来估算缺失值。DeepImpute(深度神经网络Imputation)将基因分为目标基因(待估算的基因)和训练基因(与目标基因高度相关,用于训练神经网络以确定数据分布),用于模型训练。Zhou等人利用迁移学习从DNA甲基化数据中估算缺失的表达值,并开发了一种方法,即所谓的TDimpute。在scIGANs中,基因表达矩阵被划分为小图像,插补过程被视为图像恢复的过程。Wang等人开发了scGNN(单细胞图神经网络),使用图神经网络来学习细胞-细胞关系,并结合三个自动编码器来估算“dropout”事件和细胞聚类。
尽管已经提出了大量的插补方法,并且大多数插补方法在不同的场景中都取得了良好的性能,但仍缺乏对最先进插补方法性能的全面比较。此外,还需要通过更全面的实验来改进比较。在这项研究中,作者在以下方面对六个模拟数据集和两个真实的scRNA-seq数据集上总共12种可用的插补方法进行了评估和比较。
- 首先,作者研究了现有方法恢复真实基因表达分布的能力。
- 其次,作者评估了细胞聚类时在区分不同细胞类型方面的性能。
- 第三,作者分别通过bulkRNAseq数据和scRNA-seq数据预测的差异表达基因的重叠来测试现有方法检测的差异表达基因。
- 最后,作者评估了这些方法重建细胞轨迹的能力。
最重要的是,建立了scIMC(单细胞插补方法比较平台),这是第一个计算平台,允许感兴趣的研究人员在其定制的数据集上对最先进的插补方法进行数据插补和下游比较分析,并提供可视化结果分析,以找出哪种方法最适合特定下游任务中的数据集。
使用方法
在这项工作中,作者构建了一个无偏的框架,以定量评估和比较现有的最先进的scRNA-seq数据插补方法。基于这一框架,在六个模拟数据集和两个真实数据集上比较了插补方法在多种广泛使用的指标方面的性能。基准测试框架的总体概述如图1所示。可以看出,涉及三个主要步骤:A数据预处理、B缺失值插补,C下游比较分析,详细描述如下 :
数据预处理:在六个模拟数据集和两个真实数据集上进行了基准测试。对于每个数据集,通常分两个子步骤对原始基因表达矩阵(插补前)进行预处理(见图1A)。首先,对矩阵进行归一化,以便将需要处理的数据(通过某种算法)限制在一定范围内。标准化是为了方便后续的数据处理,以及确保程序运行过程中更快收敛。然后,对归一化矩阵进行对数变换。log转换旨在更方便地找到数据之间的关系(可以理解为更好的数据可视化),使数据的呈现接近我们想要的假设,从而更好地进行统计推断。
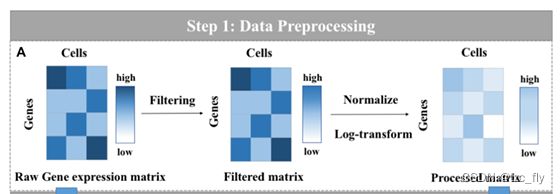
- 图1A:所有数据集都是通过去除在少于两个细胞中表达的基因来过滤掉的,这些基因被称为低表达基因。通过“scanpy”对数据集进行标准化。然后,对归一化矩阵进行对数变换。
缺失值插补:表1总结了专门为scRNA-seq数据插补设计的21种最先进的插补方法,其中有11种基于模型的方法和10种基于深度学习的方法。如表1所示,基于模型的方法可以根据其使用的信息进一步分为三个子类,例如跨细胞的信息、跨基因的信息以及跨细胞和基因的信息。基于深度学习的方法可以根据其深度网络类型进一步划分,包括自动编码器、MLP、图神经网络和其他网络。作者试图实现所有的方法,但其中只有12种成功执行,分别是SAVER、scTSSR、MAGIC、scImpute、DrImpute、scNPF、AutoImpute、ALRA、DCA、DeepImpute、scGNN和scIGAN。

- 图1B:插补方法主要分为两类:基于模型的方法,基于深度学习的方法。

- 在该表中,Cell and Gene-based是指使用跨细胞和基因的信息的方法,Cell-based是使用跨细胞的信息的方式,Gene-based是使用跨基因信息的方式。此外,Auto-based是基于自动编码器的方法,MLP-based表示基于多层感知器的方法,Graph-based是指基于图网络的方法,Other-based表示了基于其他网络的方法。此外, G ∗ C G*C G∗C表示 gene ∗ * ∗cell 表达矩阵, C ∗ G C*G C∗G表示cell ∗ * ∗gene表达矩阵。 G – G G–G G–G表示gene − - −gene相互作用网络。该表中的前11种方法是基于模型的方法,其他方法是基于深度学习的方法。
下游比较分析:为了定量比较各种方法生成的估算矩阵的好坏,作者评估了恢复实际基因表达的性能。如图1C所示,下游比较分析是为了衡量它们在实际应用场景中的性能。作者进一步比较了以下三个下游分析任务中的方法,包括聚类分析、差异表达分析和细胞轨迹分析等。

-图1C:下游比较分析。
基准数据集
在这项研究中,使用了六个模拟数据集和两个真实数据集来评估不同插补方法的性能。这里有具有不同零表达率的六个模拟数据集。两个真实的数据集是人类胚胎干细胞(ESCs)数据集和Time-course scRNA-seq数据集(Single-cell RNA-seq reveals novel regulators of human embryonic stem cell differentiation to definitive endoderm)。
模拟数据集:Splatter是一个R-Bioconductor软件包,用于模拟scRNAseq数据。作者使用Splatter生成了一个包含500个细胞和1000个基因的真计数矩阵(没有dropout的矩阵)。然后设置dropout.mid参数从1到6,为了模拟零表达率分别为0.78、0.71、0.63、0.55、0.48和0.42的六个数据集。
human ESC数据集:包括6个bulkRNA-seq样本(4个用于H1胚胎干细胞,2个用于DEC)和350个scRNA-seq细胞(212个用于H1-ESC,138个用于DEC)。bulkRNA-seq数据和scRNA-seq的基因表达矩阵中的零百分比分别为14.8%和49.1%。作者使用该数据集来评估插补方法捕获差异表达基因(DEG)的能力。对于P值(P值是显著指数)<0.05的基因,认为它是差异表达的,即所谓的DEG。分别对bulkRNA-seq数据和scRNA-seq的数据进行了edgeR来检测DEG。考虑到bulkRNA-seq数据的DEG是一个黄金标准,不同方法捕获DEG的性能被定义为bulkRNA-seq数据检测到的DEG与scRNA-seq检测到的之间的重叠。
time-course scRNA-seq数据集:作者使用了来源于从H1-ESC到DEC的分化的时间过程scRNA-seq数据。该数据集由758个细胞组成,包括从H1-ESC分化为DEC后0小时的92个细胞、12小时的102个细胞、24小时的66个细胞、36小时的172个细胞、72小时的138个细胞和96小时的188个细胞。为了评估插补方法重建轨迹的性能,作者在该数据集上执行了现有的插补方法,并使用Monocle3重建轨迹。值得注意的是,在比较的方法中,DCA和scGNN由于其固有的局限性而未能在该数据集上执行。
评价指标
基因表达矩阵表示为 X X X(RMSE和PCC中的真实基因表达矩阵),而 X ^ \widehat{X} X 是估算矩阵。为了定量评估插补方法在恢复基因表达方面的性能,作者使用了两个指标,均方根误差(RMSE)和Pearson相关系数(PCC)。为了评估和比较聚类结果和基因差异表达结果,使用了五种常见的指标:归一化互信息(NMI)、调整后的兰德指数(ARI)、轮廓系数(Si分数,silhouette coefficient)、Jaccard相似系数(Jaccard)和纯度(Purity)。至于细胞轨迹的比较,作者采用了另外两个指标:伪时间排序得分(POS,pseudo-temporal ordering score)和肯德尔秩相关得分(KOR,Kendall’s rank correlation score)。
RMSE
它是测量估算矩阵和原始矩阵之间的差异,从而计算观测值和真实值之间的偏差。RMSE定义为: R M S E ( X , X ^ ) = 1 n ∑ i = 1 n ( X ^ i − X i ) 2 RMSE(X,\widehat{X})=\sqrt{\frac{1}{n}\sum_{i=1}^{n}(\widehat{X}_{i}-X_{i})^{2}} RMSE(X,X )=n1i=1∑n(X i−Xi)2
PCC
它是为了检查估算矩阵和原始矩阵之间的相关性,定义为: ρ X , X ^ = E ( X , X ^ ) − E ( X ) E ( X ^ ) E ( X 2 ) E 2 ( X ) E ( X ^ 2 ) E 2 ( X ^ ) \rho_{X,\widehat{X}}=\frac{E(X,\widehat{X})-E(X)E(\widehat{X})}{\sqrt{E(X^{2})E^{2}(X)}\sqrt{E(\widehat{X}^{2})E^{2}(\widehat{X})}} ρX,X =E(X2)E2(X)E(X 2)E2(X )E(X,X )−E(X)E(X )其中, E ( X ) E(X) E(X)表示 X X X的mean, E 2 ( X ) E^{2}(X) E2(X)是 E ( X ) E(X) E(X)的平方。
NMI
它是指两个随机变量之间的相关性。记 l a b e l label label为原始的标签, l a b e l ^ \widehat{label} label 为聚类获得的簇标签: N M I = 2 I ( l a b e l , l a b e l ^ ) H ( l a b e l ) + H ( l a b e l ^ ) NMI=2\frac{I(label,\widehat{label})}{H(label)+H(\widehat{label})} NMI=2H(label)+H(label )I(label,label )其中: I ( l a b e l , l a b e l ^ ) = ∑ a ∈ l a b e l , b ∈ l a b e l ^ p ( a , b ) l o g p ( a , b ) p ( a ) p ( b ) I(label,\widehat{label})=\sum_{a\in label,b\in\widehat{label}}p(a,b)log\frac{p(a,b)}{p(a)p(b)} I(label,label )=a∈label,b∈label ∑p(a,b)logp(a)p(b)p(a,b) H ( l a b e l ) = ∑ a ∈ l a b e l p ( a ) l o g ( p ( a ) ) H(label)=\sum_{a\in label}p(a)log(p(a)) H(label)=a∈label∑p(a)log(p(a))其中, p ( a ) , p ( b ) , p ( a , b ) p(a),p(b),p(a,b) p(a),p(b),p(a,b)分别表示样本属于簇 a a a的概率,样本属于簇 b b b的概率,样本同时属于簇 a a a和簇 b b b的概率。
ARI
它测量两个数据分布之间的一致程度。我们假设有 m m m个细胞,它们被聚类成 k k k个聚类。 { u i } i m \left\{u_{i}\right\}_{i}^{m} {ui}im表示预测的簇标签, { v j } j m \left\{v_{j}\right\}_{j}^{m} {vj}jm表示真实的簇标签。
轮廓系数
它用于评估插补方法的细胞聚类性能。它结合了内聚和分离。Si得分越接近1,聚类就越准确;越接近-1,结果越差。Si分数定义为: S i = b i − a i m a x ( b i , a i ) Si=\frac{b_{i}-a_{i}}{max(b_{i},a_{i})} Si=max(bi,ai)bi−ai其中, a i a_{i} ai表示第 i i i个样本与同一聚类中的所有其他样本之间的平均距离, b i b_{i} bi表示给定聚类中的第 i i i个样本与所有样本(不包含第 i i i个样本的簇)之间的平均间距。
Jaccard相似系数
利用Jaccard相似系数(Jaccard)来评估插补方法的基因差异的重叠度。Jaccard用于比较样本之间的相似性。Jaccard系数值越大,样本的相似性就越高。Jaccard定义为: J ( A , B ) = ∣ A ∩ B ∣ ∣ A ∪ B ∣ J(A,B)=\frac{|A\cap B|}{|A\cup B|} J(A,B)=∣A∪B∣∣A∩B∣其中 A A A和 B B B是两个集合。Jaccard是 A A A和 B B B的交并比。
纯度
这是一个常用的聚类评估指标。
伪时间排序得分
它可用于评估细胞排序性能。POS为: P O S = ∑ i = 1 n − 1 ∑ j > i g ( i , j ) POS=\sum_{i=1}^{n-1}\sum_{j>i}g(i,j) POS=i=1∑n−1j>i∑g(i,j)其中 n n n是样本的数量, g ( i , j ) g(i,j) g(i,j)是表征有序路径中的第 i i i个和第 j j j个细胞的顺序与它们基于外部信息的期望顺序匹配程度的得分。
KOR
它经常被用来衡量两个排名之间的对应程度。其定义为: τ = 4 P n ( n − 1 ) − 1 \tau=\frac{4P}{n(n-1)}-1 τ=n(n−1)4P−1其中 n n n是样本数量, P P P是两个排名在给定样本之后的样本数量之和。
额外的工具
为了进行DEG分析,作者在scRNA-seq数据上运行了edgeR,当一个基因的P值<0.05时,我们认为它是差异表达的。
Monocle3被用于重建scRNA-seq数据的细胞轨迹。作者在R3.6.3环境中使用默认参数实现了Monocle3。值得注意的是,UMAP是Monocle3的默认可视化方法,用于可视化数据的细胞轨迹。
TSCAN是一种用于重建scRNA-seq分析中的伪时间轨迹的工具(TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis.)。它通过基于聚类的最小生成树方法对细胞进行排序。
结果
基因差异表达分析
基因差异表达分析,作为另一种常见的下游分析,是指对其表达水平取决于某些变量的基因进行分析。scRNA-seq可以深入了解单个细胞中基因表达的随机性,而这些差异表达基因会影响不同细胞亚群的定义。因此,在检测不同细胞类型中的已知差异基因时,有效的插补方法应保持scRNA-seq与bulk RNA-seq的一致性。由于缺乏差异表达分析的黄金标准,作者将bulkRNA-seq数据预测的差异表达基因作为黄金参考。差异基因表达分析在Human ESCs数据集上。
值得注意的是,DCA未能在此数据集上执行,因为计数矩阵被视为已归一化。edgeR通常用于分析基因差异表达。作者对bulkRNA-seq数据和scRNA-seq的原始和插补计数矩阵进行edgeR,然后用火山图可视化结果,如图2所示,其中x轴表示log Fold Change(log FC),y轴表示−log(P Value)。由于edgeR无法根据AutoImpute的结果运行,作者只比较了其余10种方法的性能。捕获DEG的性能被定义为bulkRNA-seq数据检测到的DEG与scRNA-seq检测到的数据之间的重叠。

- 图2:基因差异表达分析比较。
从图2中可以看到,与原始matrix相比,scImpute的估算数据检测到更多的差异表达基因。与bulkRNA-seq的结果相比,scImpute和ALRA具有最相似的形状。为了直观分析,表2中列出了10种插补方法的插补数据检测到的差异表达基因的数量。原始scRNA-seq数据的零表达率远高于bulkRNA-seq的数据,并且与bulkRNA-seq数据共享的DEG最少。如图所示,我们观察到由估算数据检测到的差异表达基因的数量大于由原始scRNA-seq数据(不包括scGNN)检测到的数量。

- 表2:DEG数量统计。
轨迹分析
细胞轨迹的重建对于在scRNAseq数据中通过时间进程探索细胞周期动力学模式至关重要。细胞轨迹分析包括三个主要步骤:降维、聚类和轨迹重建。尽管在scRNA-seq数据中广泛使用各种细胞轨迹重建方法,但它们受到“dropout”事件的严重影响。作者在time-course scRNA-seq数据集上评估插补方法,使用Monocle3和TSCAN重建轨迹。
POS和KOR得分用于测量真实时间标签和伪时间标签之间的相关性。Monocle3构建的细胞轨迹如图3所示,表3列出了不同插补方法的POS和KOR。从表3中可以看出,scImpute实现了由估算数据推断的细胞轨迹与真实细胞顺序之间的最高对应,POS为0.928,KOR为0.743,并且在没有预处理步骤的TSCAN中也表现良好。scTSSR在具有预处理步骤的TSCAN中表现良好,POS为0.918,KOR为0.734。此外,SAVER、ALRA和scIGAN的POS和KOR得分较低,表明它们的结果较差。没有预处理步骤的SAVER和ALRA的POS和KOR甚至是负的。结果表明,scImpute最适合于探索scRNA-seq数据中的细胞轨迹。
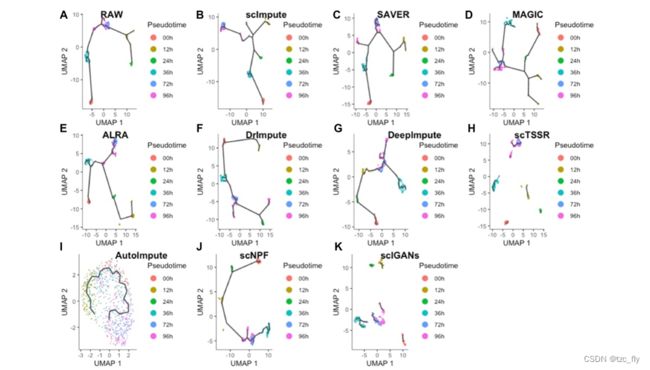
- 图3:轨迹重建结果。

- 表3:POS(w)和KOR(w)表示通过具有预处理步骤的TSCAN获得的POS和KOR分数。POS和KOR表示通过TSCAN在没有预处理步骤的情况下获得的POS和KOR得分。