文献题目:HMMRATAC: a Hidden Markov ModeleR for ATAC-seq
DOI(url): https://doi.org/10.1093/nar/gkz533
杂志:Nucleic Acids Research
发表日期:14 June 2019
关键点
本文利用 ATAC-seq 技术原理中的转座酶插入特性,设计了一种专门针对 ATAC-seq 的隐马尔科夫模型,这种半监督机器学习方法可以用来鉴定染色质开放区域。
参考意义
ATAC-seq 作为一种定位染色质开放区域的手段目前应用已经非常广泛了,因为相对易于操作目前也应用到了单细胞领域。但是目前关于ATAC-seq的流程绝大多数都是按照 ChIP-seq 流程来处理的,call peak 方法十有八九使用的都是 MACS。
ATAC-seq 利用了 Tn5 转座酶优先插入nucleosome-free regions(NFR)的特性,但是 Tn5 也有可能插入相邻核小体之间的连接区,此时其 DNA fragement 会更长(超过150bp)而且和相邻核小体的个数相关。针对双端测序数据,我们可以根据比对后的位置或插入长度来推断它们的片段长度。如果将 nucleosome free 和 mononucleosome 片段长都和频率的关系图展示出来,可以看出两者的分布不同。
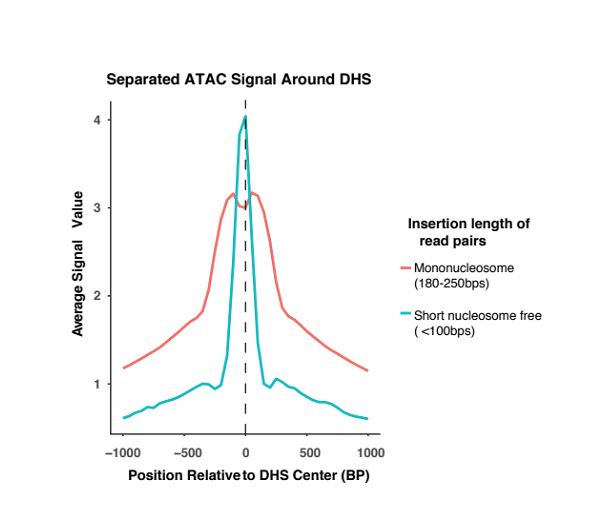
目前还没有工具可以同时考虑 ATAC-seq 中的 NFR 和核小体信息。而本文作者开发的分析工具 HMMRATAC 则采用了「分解和整合」的思路,首先把一套数据首先分解为来自于NFR 和核小体区域的不同覆盖信号层,然后在隐马尔可夫模型中学习开放染色质区域信号层之间的关系,并用于预测开放染色质。下图是一个整体的分析流程。

在文章中作者将这个工具和 MACS2 与 F-seq 进行了比较,HMMRATAC 在大多数测试中表现优于前两者。这个软件本身使用 Java 来实现的,目前作者也提到其处理速度相对较慢,是后续优化的一个重点。
软件地址:https://github.com/LiuLabUB/HMMRATAC
相关内容
顺式调控元件与反式作用因子相互作用组成了真核生物转录调控过程,这一过程与染色质核小体的动态定位相关,调控因子的结合需要裸露的无核小体的 DNA 区域,即开放的染色质位点(open chromatin)。从全基因组水平定位开放染色质位点有助于发掘基因组调控元件,进而研究基因表达调控机制。

目前使用最多的两种方法是 DNase-seq 和 ATAC-seq。
DNaseⅠ 超敏感位点 (DNase I hypersensitive sites, DHs) 测序使用限制性内切酶(DNase I)对样品进行了片段化处理。染色质开放区域的反式作用因子的结合将导致这些区域缺少核小体结构,染色质裸露、结构疏松,易于与 DNaseⅠ 结合并发生剪切 , 从而表现出对 DNaseⅠ 的高度敏感性。包括启动子、增强子、绝缘子和抑制因子等在内的多种基因调控元件均与 DHs 紧密关联。
转座子虽然对于裸露 DNA 表现出随机插入的特性,但对于染色质而言应同样表现出偏好转座的特点,即优先在开放染色质区插入染色质。转座酶可接近性 (assay for transposase accessible chromatin, ATAC) 测序就是利用了这个特性。相比于 DHs 测序,ATAT 测序操作简单快捷,检测灵敏度高,实验重复性好的特点。细胞核起始量 DHs 测序是 ATAC 测序的 3~5 个数量级。
文献题目:Essential guidelines for computational method benchmarking
DOI(url): https://doi.org/10.1186/s13059-019-1738-8
杂志:Genome Biology
发表日期:20 June 2019
关键点
计算方法基准分析的综述指南
参考意义
作为一个生物信息「调包侠」,我们平时在分析数据的时候经常会面临一个问题,这几种计算方法我究竟选哪一个?根据不完全统计,目前用来分析单细胞RNA-seq的方法已经有400多种了,这里就带来了一个问题,选择不同的方法通常会带来不同甚至是很不同的结果,我们该如何选择。这篇文章作者总结了进行高质量基准分析(computational method benchmarking)的关键指南和建议。下图为指南内容的概括。

这上面十点要注意的指南中,首先是定义分析的目的和范围,比如有一类基准分析是有开发者本身使用的,他们的目的是证明自己方法的优势;也有通过系统比较一系列方法进行中立性评价分析的。中立的基准测试应该尽可能全面,同时测试也应该充分和原开发者沟通以便在最佳性能的前提下进行测试。在任何情况下都应该避免因为特别关注某一种方法带来的偏差。而针对新方法优点的评价应该仔细设计评价的标准,一个常见的问题是使用竞争方法的默认参数,然后不停的调整自己方法的参数。
关于上述10个原则对于一个优秀基准的“多么重要”,以及与每个原则相关的关键和潜在问题,作者总结了如下一个表格;
| Principle | How essential | Tradeoffs | Potential pitfalls |
|---|---|---|---|
| 1. Defining the purpose and scope | +++ | How comprehensive the benchmark should be | Scope too broad: too much work given available resourcesScope too narrow: unrepresentative and possibly misleading results |
| 2. Selection of methods | +++ | Number of methods to include | Excluding key methods |
| 3. Selection (or design) of datasets | +++ | Number and types of datasets to include | Subjectivity in the choice of datasets: e.g., selecting datasets that are unrepresentative of real-world applicationsToo few datasets or simulation scenariosOverly simplistic simulations |
| 4. Parameter and software versions | ++ | Amount of parameter tuning | Extensive parameter tuning for some methods while using default parameters for others (e.g., competing methods) |
| 5. Evaluation criteria: key quantitative performance metrics | +++ | Number and types of performance metrics | Subjectivity in the choice of metrics: e.g., selecting metrics that do not translate to real-world performanceMetrics that give over-optimistic estimates of performanceMethods may not be directly comparable according to individual metrics (e.g., if methods are designed for different tasks) |
| 6. Evaluation criteria: secondary measures | ++ | Number and types of performance metrics | Subjectivity of qualitative measures such as user-friendliness, installation procedures, and documentation qualitySubjectivity in relative weighting between multiple metricsMeasures such as runtime and scalability depend on processor speed and memory |
| 7. Interpretation, guidelines, and recommendations | ++ | Generality versus specificity of recommendations | Performance differences between top-ranked methods may be minorDifferent readers may be interested in different aspects of performance |
| 8. Publication and reporting of results | + | Amount of resources to dedicate to building online resources | Online resources may not be accessible (or may no longer run) several years later |
| 9. Enabling future extensions | ++ | Amount of resources to dedicate to ensuring extensibility | Selection of methods or datasets for future extensions may be unrepresentative (e.g., due to requests from method authors) |
| 10. Reproducible research best practices | ++ | Amount of resources to dedicate to reproducibility | Some tools may not be compatible or accessible several years later |
相关内容
这篇文章的作者最近才在 NBT 发布一篇单细胞分析方法的「测评」A comparison of single-cell trajectory inference methods。只能说特别优秀。
文献题目:Moving beyond P values: data analysis with estimation graphics
DOI(url): https://doi.org/10.1038/s41592-019-0470-3
杂志:Nature Methods
发表日期:19 June 2019
关键点
除了P值还应该做点什么
参考意义
这篇文章介绍了应该如何分析两组数据是否有显著性差异,除了 P 值,我们还应该展示些什么。

如上图所示,用星号标记的条形图仅显示均值和误差,掩盖了具体的观察值,箱形图同样不显示复杂属性(例如,双峰)和单个观察值。另外,条形图和箱形图都是只展示最终的P值计算结果但是没有展示 null 分布(H0 时样本的分布)本身。另外,可以使用每个数据具体的点图来展示数据。当然更好的方法就是使用坐着推荐的图 e。也就是采用估算统计的方法对数据进行展示(Estimation statistics),它使用熟悉的统计概念:均值,均值差(两个不同组中的平均值之间的绝对差异)和误差线。侧重于关注实验的效应值 ,而不是由P值产生的错误二分法。从图 e 可以看出其首先将所有数据点都以swarmplot的形式呈现,并且尽量展示原数据的分布。同时添加一个独立但是和原始坐标轴对应的坐标轴显示均值差和效应值。

相关内容
R 包地址:https://github.com/ACCLAB/dabestr
文献题目:TransLiG: a de novo transcriptome assembler that uses line graph iteration
DOI(url): https://doi.org/10.1186/s13059-019-1690-7
杂志:Genome Biology
发表日期:23 April 2019
关键点
比Trinity更厉害的转录本组装工具
参考意义
TransLiG 是第一个通过phasing和收缩路径将双端测序信息和测序深度信息整合到从头组装的方法。通过评估,TransLiG 比已有的转录本从头组装工具在 accuracy computing resources 都有很大的优势。
使用 6 种比对方法在三种真实数据集中分析灵敏度 sensitivity

使用 6 种比对方法在三种真实数据集中分析精确度 precision

比较CPU time

比较 RAM 使用情况

作者也分析了TransLiG 具有优势的几个原因:
- Firstly, TransLiG constructs more accurate splicing graphs by reconnecting fragmented graphs via iterating different lengths of smaller k-mers.
- Secondly, TransLiG substantially integrates the sequence depth and paired-end information into the assembling procedure via enforcing each pair-supporting path being included in at least one assembled transcript.
- Thirdly, TransLiG accurately links the in-coming and out-going edges at each node via iteratively solving a series of quadratic programmings, which are optimizing the utilizations of the paired-end and sequencing depth information.
- Finally, TransLiG benefits from the iterations of weighted line graphs constructed by repeatedly phasing transcript-segment-representing paths.
相关内容
如果再有转录组拼接的需求,我会用它试一试。文中提到的bridger 我曾经做过比较详细的测试,这个包目前已经不再维护了,而且安装的时候有一个bug需要手动修改源码。SOAPdenovo-trans 我也使用过,确实在速度和内存控制上要比 trinity 好很多,而且结果也没有多差。
https://sourceforge.net/projects/transcriptomeassembly/files/
https://zenodo.org/record/2576226
除了以上内容之外还有一篇 gene set enrichment analysis 分析相关的文章:Towards a gold standard for benchmarking gene set enrichment analysis
