1、 什么是苯丙酮尿症?
苯丙酮尿症(PKU)是一种遗传代谢病,由于基因突变导致肝脏中苯丙氨酸羟化酶缺乏,引起苯丙氨酸代谢障碍,苯丙氨酸及代谢产物在血液中浓度增加,严重影响神经系统发育,使大脑发育受到损伤最后导致智力低下。
2,为什么会发生苯丙酮尿症?
苯丙酮尿症是父母遗传给孩子的,这种病的遗传方式在医学上叫做常染色体隐形遗传,患儿父母外表是“正常人”,但都是致病基因的携带者,只有父母双方为致病基因携带者时,才有可能生育患儿。这样的父母每次怀孕都有四分之一的几率生育苯丙酮尿症患儿。
3、 苯丙酮尿症是怎样引起神经系统损伤的?
苯丙氨酸是人体生长和代谢的必需氨基酸,食物中的苯丙氨酸进入人体后,部分合成蛋白质,一部分在苯丙氨酸羟化酶作用下转变为酪氨酸。当苯丙氨酸羟化酶缺乏,正常转化的酪氨酸减少,会导致多巴胺肾上腺素减少,影响神经系统的发育。苯丙氨酸羟化酶缺乏,正常代谢阻断,异常代谢生成苯丙酮酸增加,直接导致神经系统损伤。
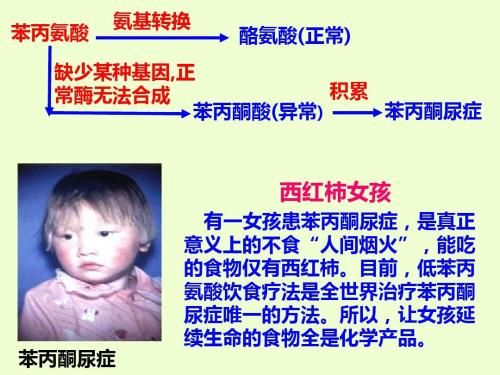
4、 苯丙酮尿症有哪些表现?
宝宝出生时外表正常,有些患儿可能有喂养困难、呕吐、睡眠不安、易哭闹等。未经治疗的患儿在生后3-4个月内逐渐出现智力发育落后,头发由黑变黄、皮肤白、汗和尿液有特殊的鼠尿臭(恶臭味)、常有皮肤湿疹。随年龄增大,病儿智力低下越明显,多数患儿有严重智力障碍,部分患儿有癫痫发作。

5、 怀疑苯丙酮尿症需要做哪些检查?
如果宝宝经新生儿疾病筛查苯丙酮尿症一项异常或宝宝出现上述表现,应做如下检查,以便尽早确诊。①血苯丙氨酸测定。②尿碟呤谱分析。③红细胞二氢生物碟呤还原酶活性测定(DHPR测定)。④基因检测,能准确诊断并鉴别分型(父母及宝宝均作)。
6、 怎样才能早期诊断苯丙酮尿症?
由于患儿早期症状不明显,必须借助实验室检测。宝宝出生后接受新生儿疾病筛查是目前最安全、简便、有效的早期发现苯丙酮尿症的方法。
7、 得了苯丙酮尿症应该怎么治疗?
低苯丙氨酸饮食治疗是目前治疗苯丙酮尿症最有效的方法,一旦确诊后,应尽快给予低苯丙氨酸饮食,严格控制蛋白质,尤其是动物蛋白质的摄入。应保证热量、蛋白质、及一定量的苯丙氨酸的食入。还要注意维生素、微量元素的补充。尽可能给苯丙酮尿症患儿的早期康复干预。
8、 为什么苯丙酮尿症患儿不能吃普通饮食,而只能吃特殊饮食?
由于普通食物中含有较高的苯丙氨酸,而体内缺乏苯丙氨酸羟化酶的患儿不能正常代谢他们,如果吃普通食物就会导致患儿智力发展落后。所以他们只能吃特殊配制的不含或少含苯丙氨酸的饮食才能健康成长。
9、 苯丙酮尿症患儿怎样进行食物选择?
目前有国产和进口的无苯丙氨酸或低苯丙氨酸的配方营养粉(俗称特殊奶粉或治疗奶粉),还有无苯丙氨酸或低苯丙氨酸的米、面粉、饼干、面条等,可以选择。天然食物中母乳是最好的低苯丙氨酸食品、蔬菜、水果、瓜类、脂类等使可以自由食用的。乳类、豆类、谷类含有一定量的苯丙氨酸是应当控制食入的食品,肉类、鱼类、蛋类是优质蛋白质可在医生指导下少量食用。
10、 丙酮尿症在治疗中家长应注意什么?
家长要做到:配合医生,坚持治疗,定期检查血苯丙氨酸浓度,不能中断,这是治疗成功的关键。只有规范的治疗、合理的饮食、稳定的血苯丙氨酸浓度,苯丙酮尿症患儿就可能与同龄儿童一样健康成长。(市妇幼保健院新生儿疾病筛查中心经筛查确诊的首例苯丙酮尿症已经在一所重点中学读高中二年级)
11、 苯丙通尿症患儿需要治疗多长时间?
苯丙酮尿症患儿的饮食治疗应越早越好,低苯丙氨酸饮食治疗至少持续到青春发育成熟期,提倡终身治疗。青春发育成熟期后如果随意进食,苯丙氨酸浓度过高也会对精神情绪有一定影响。
12、 苯丙酮尿症患儿以后如何?
苯丙酮尿症患儿若不治疗或晚治疗,大都有智力发育落后,严重的成为“痴呆儿”。通过新生儿疾病筛查,苯丙酮尿症患儿能早期发现并及时正规治疗。可使患儿智力达到正常同龄儿童,只有少数患儿治疗不配合或病情严重,血苯丙氨酸浓度控制不好,可导致智力发育落后。
13、 如何预防苯丙酮尿症?
首先要避免近亲结婚。如果父母已生育过一个苯丙酮尿症患儿?再生育必须做产前诊断。对有家族史的孕妇应采取产前诊断技术,对其胎儿是否患有苯丙酮尿症进行产前诊断。
参考文献:
National Institutes of Health Consensus Development Panel. National Institutes of Health Consensus Development Conference Statement: phenylketonuria: screening and management, October 16-18, 2000. Pediatrics 2001; 108:972.
Hofman KJ, Steel G, Kazazian HH, Valle D. Phenylketonuria in U.S. blacks: molecular analysis of the phenylalanine hydroxylase gene. Am J Hum Genet 1991; 48:791.
Erlandsen H, Stevens RC. The structural basis of phenylketonuria. Mol Genet Metab 1999; 68:103.
Fölling A. Über Ausscheidung von Phenylbrenztraubensäure in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillität. Zschr Physiol Chem 1934; 227:169.
Scriver CR, Kaufman S. The hyperphenylalaninemias. Phenylalanine hydroxylase deficiency. In: The metabolic and molecular bases of inherited disease, 8th ed, Scriver CR, Beaudet AL, Sly WS, Valle D (Eds), McGraw-Hill, New York 2001. p.1667.
Ushakova GA, Gubkina HA, Kachur VA, Lepekhin EA. Effect of experimental hyperphenylalaninemia on the postnatal rat brain. Int J Dev Neurosci 1997; 15:29.
Kienzle Hagen ME, Pederzolli CD, Sgaravatti AM, et al. Experimental hyperphenylalaninemia provokes oxidative stress in rat brain. Biochim Biophys Acta 2002; 1586:344.
Infante JP, Huszagh VA. Impaired arachidonic (20:4n-6) and docosahexaenoic (22:6n-3) acid synthesis by phenylalanine metabolites as etiological factors in the neuropathology of phenylketonuria. Mol Genet Metab 2001; 72:185.
Glushakov AV, Dennis DM, Morey TE, et al. Specific inhibition of N-methyl-D-aspartate receptor function in rat hippocampal neurons by L-phenylalanine at concentrations observed during phenylketonuria. Mol Psychiatry 2002; 7:359.