RNA-seq与miRNA-seq联合分析
RNA-seq miRNA-seq联合分析
背景知识
肝癌细胞经常会入侵门静脉系统,从而导致门静脉癌栓,但是还没有一个详尽的研究来讨论其中的作用机制,因此需要对肝癌组织(tumor),门静脉组织(PVTT),癌旁组织(normal)进行采样分析。
数据来源
数据来源于2017年5月24日清华大学更新的miRNA-seq,DNA methylation, CNV, RNA-seq
项目标题:The molecularlandscape of hepatocellular carcinoma with portal vein tumor thrombosis
实验设计:
提取了来自20个中国肝癌患者的肿瘤组织,门静脉组织和癌旁组织,共计60个样本,分别对其进行miRNA-seq,甲基化分析,拷贝数变异分析和RNA-seq分析。
数据下载网址:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE77276
RNA-seq数据分析
数据预处理
由于此数据原始数据sra太大,没有表达矩阵,提供了测序序列reads经过标准化以后,在每个基因上的数目(normalized_count),将各个样本reads count文件合并就可以得到表达矩阵。
差异表达基因筛选
根据文献所述,使用R包DESeq2筛选差异表达基因,DESeq2使用负二项分布产生的线性模型,具体原理可见如下网址
http://www.bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html#theory-behind-deseq2
分组方式:源数据为肝癌组织(tumor),门静脉组织(PVTT),癌旁组织(normal),然而由于门静脉组织也属于病变组织的一种,可以和tumor划分为一类
最终在pvalue<0.001的条件下筛选出5676个差异表达基因,具体可以参见文件condition_treated_results.txt。
聚类热图
对前20个差异表达基因绘制聚类热图,可以发现normal和tumor明显分开,这说明DESeq2找出来的差异表达基因还是蛮不错的。
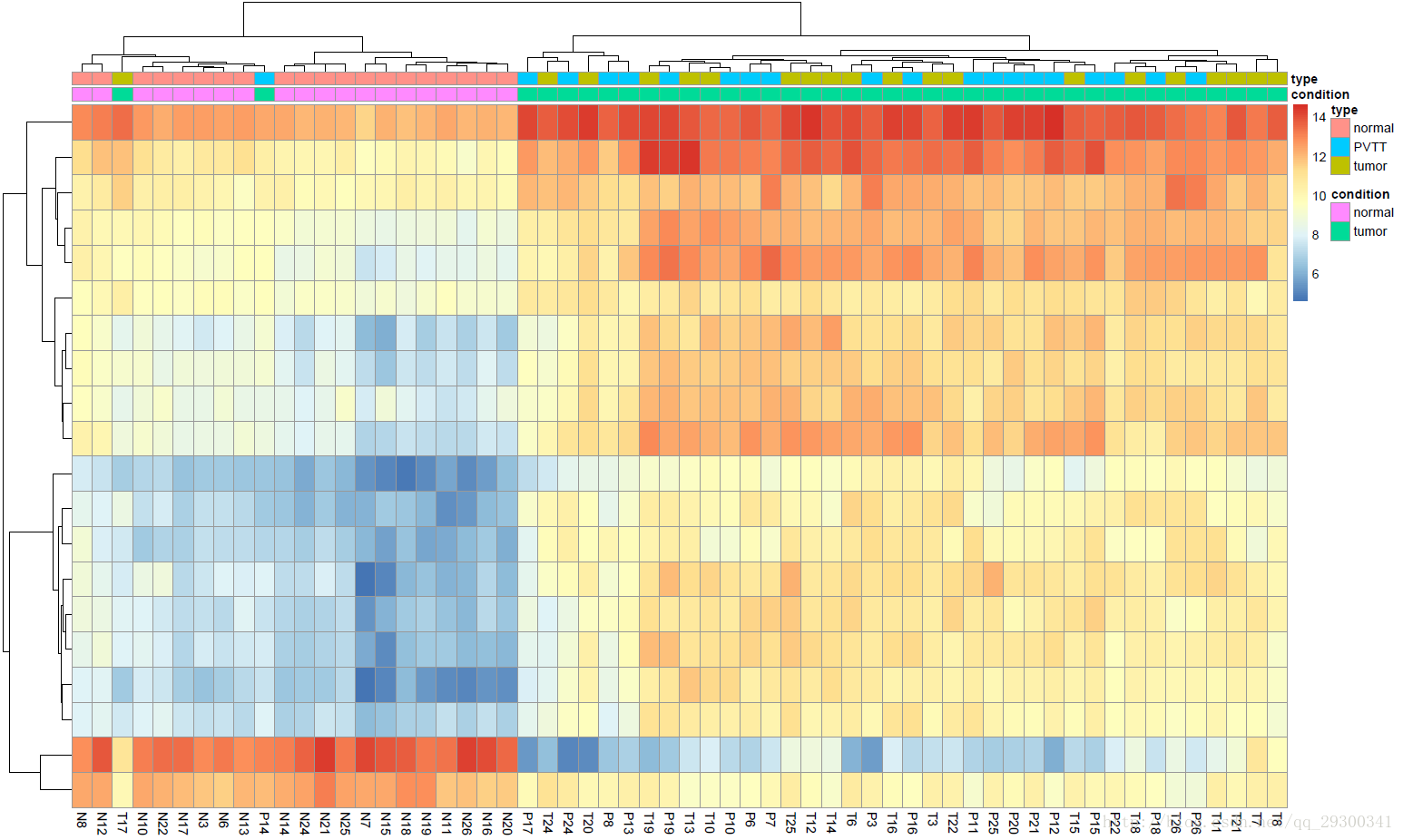
图表 1聚类热图
深度分析
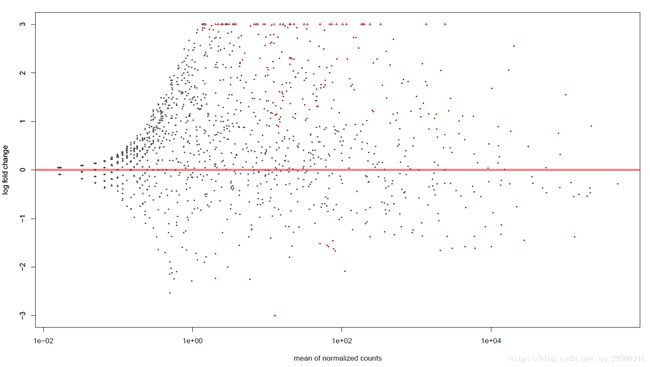
为了进一步探索数据和结果,绘制MA-plot,横坐标为每个基因上reads的数目(标准化后);纵坐标为log2fold change,即变化的程度;每个点就是一个基因,红色小点为pvalue<0.001的基因;只绘制了log2foldchange在(-3,+3)以内的基因,即改变程度在(0.125,8)倍的基因,对于不在此范围内的基因,用三角形的标志画在边界线上。

图表 2MA-plot
可以从图中看出来,黑色部分大致形成一个三角形,而红色部分(差异表达基因)包裹在黑色三角形外围。这说明用DESeq2的负二项分布模型找出来的差异表达基因,大部分都是reads数目多(测序深度高),且表达量差异很大的基因。
接下来绘制某一基因在不同组织的表达量。选取p值最小的那个基因
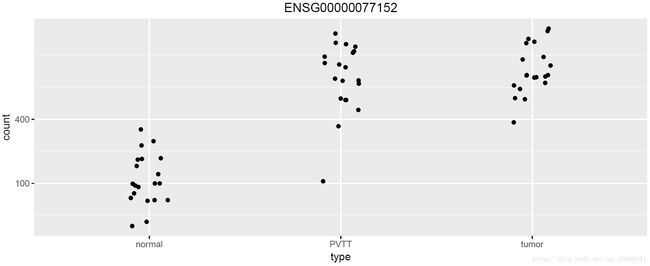
图表 3某一基因的表达量
PVTT和tumor在差异表达基因筛选的时候合为两组,此时绘图的时候仍然将它们分开。可以看到ENSG00000077152在normal和PVTT+tumor间表达量明显不同。
再接下来可以进行主成分分析,对整个表达矩阵计算主成分,然后选取前面两个主成分绘制PCA图,可以看见PCA1代表了原本36%的信息量,PCA2代表了原本10%的信息量,然后normal和其他两类比较能分得开,比起之前那次作业芯片数据,这次的紧致性要好得多。

图表 4PCA图
miRNA-seq数据分析
数据处理过程和上面的RNA-seq一样,把代码切换一下目录就成。
在2578个miRNA中,共有199个差异表达(pvalue<0.001),绘制MA-plot发现上调的居多,
图表 5MA-plot
接下来,也对差异表达的部分做了聚类热图,发现对于差异表达的部分,两组确实分得挺开的。

图表 6聚类热图
接下来也挑了p值最小的miRNA绘制reads count图,发现两组之间的差异确实蛮明显的。

图表 7p值最小miRNA
最后,进行了主成分分析,绘制PCA图,紧致性不如上面的RNA-seq,应该是前两个PCA代表的信息太少的缘故,第一主成分只有代表源数据19%的信息,第二主成分代表17%的信息,俩主成分加起来才有刚刚一个主成分那么多信息(RNA-seq第一主成分就有36%)。

图表 8PCA图
联合分析
MAGIA(miRNA和基因整合分析)是一个进行靶预测、miRNA和基因表达数据整合分析的新的网络工具。接下来,使用magia进行miRNA与基因相互作用的联合分析。
网址:http://gencomp.bio.unipd.it/magia/analysis/
Step1
由于miRNA-seq和RNA-seq是来源相同的配对数据,而且样本数有60个。联合分析算法选择MATCHED:Mutual Information
MATCHED: Mutual Information: a classicinformation measure quantifying the mutual dependence of variables, includingnon-linear relationships. Suitable for large sample size (>20 needed).
Step2
接下来的预测方式选择Pita和miRanda的交集
Pita score filter:-10 Miranda score filter:500(都是默认值)
Step3
接下来将上面分析出来的差异表达矩阵分别上传,分析即可。下面就是绘制出来的相互作用网络图。

图表 9相互作用网络
红色三角形为miRNA,绿色圆形为基因。
红色圈圈是看上去连线比较多的几个miRNA,比较重要,名字分别是:hsa-miR-760、hsa-miR-1303 、hsa-miR-671-5p、hsa-miR-324-3p、hsa-miR-423-3p
还能做出来相互作用(interaction)的程度,下载为tsv文件,
就是一张包含了MicroRNA、Gene Symbol、MutualInformation的表,Mutual Information指互信息,是信息论里一种有用的信息度量,它可以看成是一个随机变量中包含的关于另一个随机变量的信息量,或者说是一个随机变量由于已知另一个随机变量而减少的不肯定性。
也就是说,在这里MutualInformation就可以看做两者的相关程度。
就比如在下图表的截图中可以看出来,hsa-mir-1303和其对应的靶基因DBF4B、hsa-mir-501-5p和其对应的靶基因KIF2C就有很强的相关性。

图表 10相互作用表
GO注释
使用Gene Ontology官网上的在线注释功能即可,输入刚刚相互作用网络interactions.tsv文件中的基因名,进行biologicalprocess(生化反应),molecularfunction(分子功能),cellularcomponent(细胞定位)三方面的富集分析,通过富集分析可以找出在统计上显著富集的GO Term,这些富集的条目有可能与研究的目前有关。
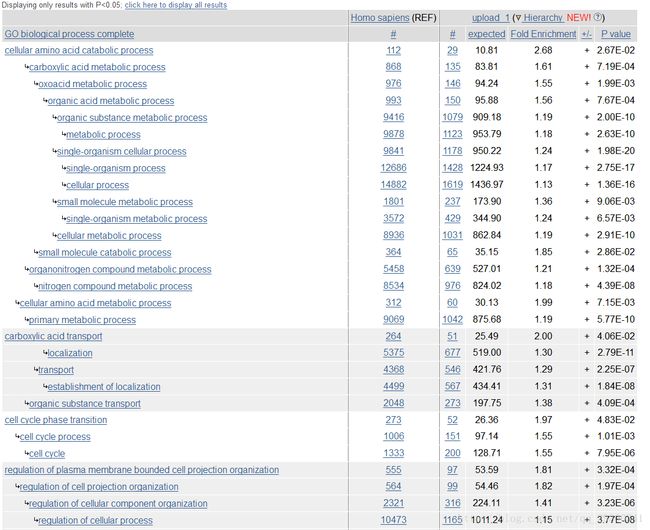
图表 11biological process
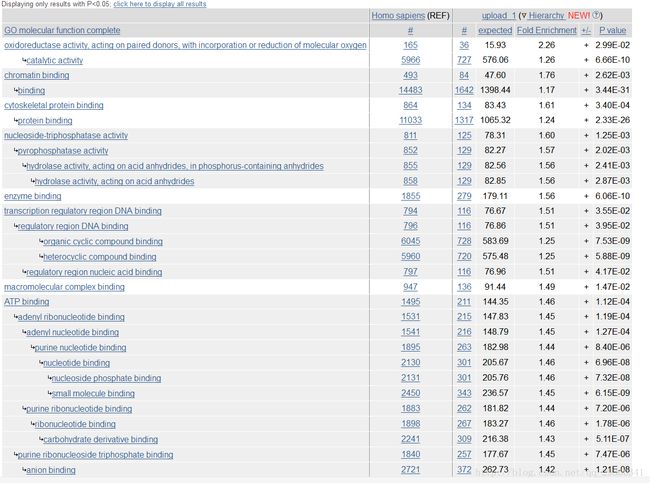
图表 12molecular function
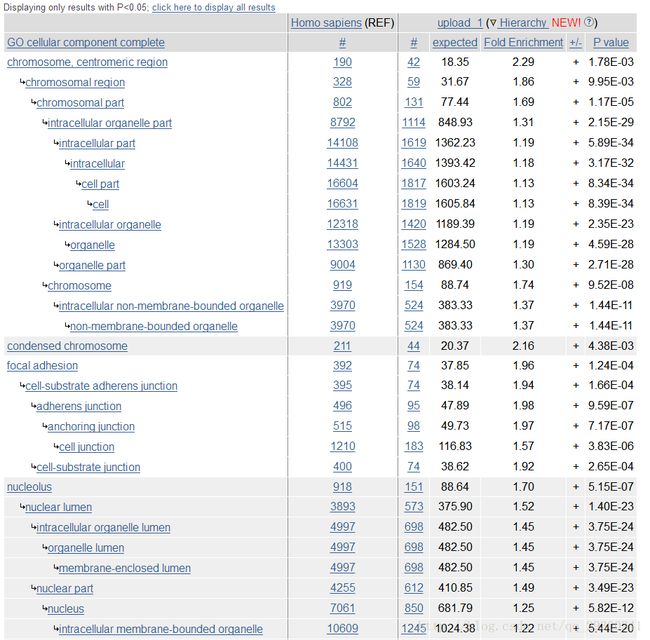
图表 13cellular component
看上去确实有一些相关的富集条目,比如分子功能:染色体绑定(chromatin binding);生化过程:有丝分裂过程(mitotic cell cycle);细胞定位:染色体部位(chromosomal part),这些都和癌症细胞的产生有着重要关系。
结语
本次实验使用的是配对的miRNA和mRNA表达谱文件,这给了我们一个通过生物信息学工具构建miRNA-mRNA相互作用网络的好机会,在系统层次的分析表明,我们找到了许多的重要miRNA和mRNA,这些对于肝癌起始和发展的过程中起着重要作用。这个全局的“miRNA-mRNA相互作用网络”对于筛选miRNA靶基因和发现新的治疗靶标有着重要意义。