我们先加载数据,数据分别来自TCGAbiolinks(直接下载自TCGA-GDC)和UCSCXena,单看临床数据。
library(TCGAbiolinks)
library(dplyr)
library(DT)
library(stringr)
library(SummarizedExperiment)
rm(list = ls())
load('E:/TCGA/TCGAbiolinks/rdata/step1-HNSC-clinical-rnaseq.rdata')
load('E:/TCGA/UCSCXena/Project/Data/HNSC/meth.HNSC.rdata')
其中Xena的数据重复计算了癌旁样本。
> dim(clinical.biolink)
[1] 528 42
> dim(clinical.Xena)
[1] 604 141
>
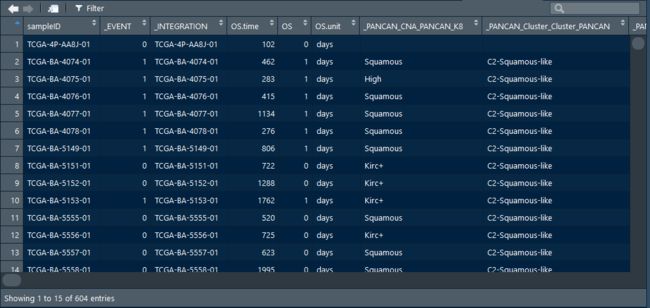

我们把clinical.Xena癌旁样本剔除掉:
> clinical.Xena$sampleID
[1] "TCGA-4P-AA8J-01" "TCGA-BA-4074-01" "TCGA-BA-4075-01" "TCGA-BA-4076-01" "TCGA-BA-4077-01" "TCGA-BA-4078-01"
[7] "TCGA-BA-5149-01" "TCGA-BA-5151-01" "TCGA-BA-5152-01" "TCGA-BA-5153-01" "TCGA-BA-5555-01" "TCGA-BA-5556-01"
[13] "TCGA-BA-5557-01" "TCGA-BA-5558-01" "TCGA-BA-5559-01" "TCGA-BA-6868-01" "TCGA-BA-6869-01" "TCGA-BA-6870-01"
其中14-15位01的是原发肿瘤样本,06是复发癌,11是癌旁。我们做一个统计看看:
> clinical.Xena$sampleID %>% word(sep = '-', 4) %>% table()
.
01 06 11
528 2 74
> ind = (clinical.Xena$sampleID %>% word(sep = '-', 4)) == '01'
> ind %>% table()
.
FALSE TRUE
76 528
> clinical.Xena = clinical.Xena[ind, ]
528例原发肿瘤样本,和2例复发肿瘤,74例癌旁正常样本。
对行名进行赋值:
> rownames(clinical.biolink) = clinical.biolink$submitter_id
> rownames(clinical.Xena) = clinical.Xena$sampleID
> rownames(clinical.Xena)[1:5]
[1] "TCGA-4P-AA8J-01" "TCGA-BA-4074-01" "TCGA-BA-4075-01"
[4] "TCGA-BA-4076-01" "TCGA-BA-4077-01"
> rownames(clinical.biolink)[1:5]
[1] "TCGA-4P-AA8J" "TCGA-BA-4074" "TCGA-BA-4075" "TCGA-BA-4076"
[5] "TCGA-BA-4077"
> rownames(clinical.biolink) %>%
substr(., 1, 12) -> rownames(clinical.biolink)
> rownames(clinical.Xena)[1:5]
[1] "TCGA-4P-AA8J" "TCGA-BA-4074" "TCGA-BA-4075" "TCGA-BA-4076"
[5] "TCGA-BA-4077"
貌似行名一致。。。。
我们看看生存事件:
> clinical.biolink$vital_status %>% table()
.
alive dead
304 224
> clinical.Xena$OS %>% table()
.
0 1
302 223
Xena的数据死亡发生例数少1个,存活例数少2个。总数少3个(=528-302-223),仔细看是NA值:

我们把他们的样本名调出来, 看看Biolinks的数据是怎样的:
> rownames(clinical.Xena)[is.na(clinical.Xena$OS)]
[1] "TCGA-BB-4224" "TCGA-CQ-A4CA" "TCGA-H7-A6C4"
> samples.na = rownames(clinical.Xena)[is.na(clinical.Xena$OS)]
> clinical.biolink[samples.na, ]

我们看看官网的资料,是否与TCGAbiolinks下载的数据一致:

这个是缺失资料的更新。
我们接着看:
> clinical.biolink$tissue_or_organ_of_origin %>% table() %>% sort(decreasing = T)
.
Tongue, NOS
130
Larynx, NOS
116
Overlapping lesion of lip, oral cavity and pharynx
70
Floor of mouth, NOS
54
Tonsil, NOS
46
Base of tongue, NOS
24
Mouth, NOS
23
Cheek mucosa
19
Hypopharynx, NOS
9
Oropharynx, NOS
9
Gum, NOS
8
Hard palate
4
Lip, NOS
3
Anterior floor of mouth
2
Lower gum
2
Border of tongue
1
Mandible
1
Palate, NOS
1
Pharynx, NOS
1
Posterior wall of oropharynx
1
Retromolar area
1
Supraglottis
1
Upper Gum
1
Ventral surface of tongue, NOS
1
Overlapping lesion of lip, oral cavity and pharynx项太长,重新命名:
> ind = clinical.biolink$tissue_or_organ_of_origin==
+ 'Overlapping lesion of lip, oral cavity and pharynx'
> clinical.biolink$tissue_or_organ_of_origin[ind] = 'Oral, lip and pharynx'
> clinical.Xena$anatomic_neoplasm_subdivision %>% table() %>% sort(decreasing = T)
.
Oral Tongue Larynx Oral Cavity Floor of mouth Tonsil
133 117 73 63 45
Base of tongue Buccal Mucosa Alveolar Ridge Hypopharynx Oropharynx
27 23 18 10 9
Hard Palate Lip
7 3
> clinical.biolink$tissue_or_organ_of_origin %>% table() %>% sort(decreasing = T)
.
Tongue, NOS Larynx, NOS
130 116
Oral, lip and pharynx Floor of mouth, NOS
70 54
Tonsil, NOS Base of tongue, NOS
46 24
Mouth, NOS Cheek mucosa
23 19
Hypopharynx, NOS Oropharynx, NOS
9 9
Gum, NOS Hard palate
8 4
Lip, NOS Anterior floor of mouth
3 2
Lower gum Border of tongue
2 1
Mandible Palate, NOS
1 1
Pharynx, NOS Posterior wall of oropharynx
1 1
Retromolar area Supraglottis
1 1
Upper Gum Ventral surface of tongue, NOS
1 1
> clinical.Xena$anatomic_neoplasm_subdivision %>% table() %>% sort(decreasing = T)
.
Oral Tongue Larynx Oral Cavity Floor of mouth Tonsil
133 117 73 63 45
Base of tongue Buccal Mucosa Alveolar Ridge Hypopharynx Oropharynx
27 23 18 10 9
Hard Palate Lip
7 3
我们可以清晰的看出,很多不一样的。看看具体什么不一样:
clinical.biolink = clinical.biolink[rownames(clinical.Xena),]#确保行一致
clinical.merge = data.frame(
subdivision.Xena = clinical.Xena$anatomic_neoplasm_subdivision,
subdivision.biolink = clinical.biolink$tissue_or_organ_of_origin,
row.names = rownames(clinical.Xena))

我们以舌癌(tongue cancer)为例,找出不一致的样本:
> tongue.Xena = grepl('[Tt]ongue', clinical.merge$subdivision.Xena)
> tongue.biolink = grepl('[Tt]ongue', clinical.merge$subdivision.biolink)
> tongue.Xena %>% table()
.
FALSE TRUE
368 160
> tongue.biolink %>% table()
.
FALSE TRUE
372 156
TCGAbiolink中少了4例。
具体看看:
> clinical.merge[tongue.Xena != tongue.biolink, ]
subdivision.Xena subdivision.biolink
TCGA-BA-5557 Oral Tongue Oral, lip and pharynx
TCGA-BA-A4IG Base of tongue Oropharynx, NOS
TCGA-BB-7872 Oral Tongue Mouth, NOS
TCGA-CN-6016 Floor of mouth Tongue, NOS
TCGA-CN-A642 Floor of mouth Tongue, NOS
TCGA-CQ-6218 Floor of mouth Tongue, NOS
TCGA-CQ-6225 Oral Tongue Oral, lip and pharynx
TCGA-CQ-7067 Oral Tongue Mouth, NOS
TCGA-CQ-7072 Floor of mouth Tongue, NOS
TCGA-D6-A6EM Oral Tongue Mouth, NOS
TCGA-MZ-A6I9 Base of tongue Oropharynx, NOS
TCGA-T2-A6WX Oral Tongue Floor of mouth, NOS
528例中有12例的舌癌诊断记录是有差别的。我们看看最新官网的:



我们发现TCGAbiolinks下载的数据与官网的数据是一致的。
再来看看数据库介绍的文档:
UCSC-Xena
The Cancer Genome Atlas
The Cancer Genome Atlas (TCGA), a collaboration between the National Cancer Institute (NCI) and National Human Genome Research Institute (NHGRI), has generated comprehensive, multi-dimensional maps of the key genomic changes in 33 types of cancer. The TCGA dataset, describing tumor tissue and matched normal tissues from more than 11,000 patients, is publically available and has been used widely by the research community. The data have contributed to more than a thousand studies of cancer by independent researchers and to the TCGA research network publications.
Data on this hub is derived from files download from TCGA Data Coordinating Center (DCC) in Jan 2016, and Broad Firehose(analyses 2016-01-28).
bioconductor中的TCGAbiolinks
TCGAbiolinks: An R/Bioconductor package for integrative analysis with GDC data
Bioconductor version: Release (3.8)The aim of TCGAbiolinks is : i) facilitate the GDC open-access data retrieval, ii) prepare the data using the appropriate pre-processing strategies, iii) provide the means to carry out different standard analyses and iv) to easily reproduce earlier research results. In more detail, the package provides multiple methods for analysis (e.g., differential expression analysis, identifying differentially methylated regions) and methods for visualization (e.g., survival plots, volcano plots, starburst plots) in order to easily develop complete analysis pipelines.
可以看出UCSC中的数据是来自2016年的TCGA Data Coordinating Center (DCC)和Broad Firehose
而自2016年后,TCGA官网进行了多少次更新呢?